|Articles|April 1, 2011
- Special Issues-04-01-2011
- Volume 29
- Issue 4
Fast LC for Conventional HPLC Systems
Author(s)Joseph Helble
Many laboratory budgets do not allow the purchase of new ultrahigh-pressure liquid chromatography (UHPLC) systems, and workloads typically are not declining. Fast LC incorporates the use of faster mobile phase flow rates and smaller particles to achieve separations in less time and with equivalent resolution to traditional high performance liquid chromatography (HPLC).
Advertisement
Many laboratory budgets do not allow the purchase of new ultrahigh-pressure liquid chromatography (UHPLC) systems, and workloads typically are not declining. Fast LC incorporates the use of faster mobile phase flow rates and smaller particles to achieve separations in less time and with equivalent resolution to traditional high performance liquid chromatography (HPLC). Fast LC is one way to get more productivity out of existing HPLC technology and prepare for the next generation of UHPLC systems with more-efficient separation schemes.
Fast liquid chromatography (LC) has been discussed since the 1980s (1), but there had been only modest interest in it until the advent of ultrahigh-pressure liquid chromatography (UHPLC) systems. Fast LC affords fast flow rates through shorter columns, which are often packed with smaller particles. Some reasons for developing fast LC methods now include
- You can run more samples in the same time frame.
- It is where the field of high performance liquid chromatography (HPLC) is going.
- You can develop fast methods with the existing HPLC systems in your laboratory.
- You can save time and solvent by coverting to fast LC.
- You can get better resolution and equivalent quantitation to conventional HPLC.
- You can have more time to analyze your data and provide a value added dimension to your results.
By learning to develop fast LC methods, you will be better prepared to develop methods with UHPLC systems.
If fast LC saves time and money and has all of these benefits, then why do not more chromatographers adopt this technology?
I have discussed this issue with a number of colleagues and have found there are six reasons that they give for not converting their methods to fast LC. I will list them here and discuss them throughout this review:
- They do not know how to implement the technology.
- An effort to transfer an existing method to a fast LC method resulted in worse resolution.
- They simply cannot afford to lose resolution for decreased cycle time.
- The stability of the columns will not be as good as 5-µm packing.
- The need to quantitate peaks with reliable retention times and peak areas is critical.
- A method used in a GMP environment would need to be revalidated.
Although there are numerous articles explaining that fast LC is useful, there are not many that explain how to implement the technology. Even when there is an explanation, some of the significant elements of fast LC are ignored. Most discussions of fast LC begin with band-broadening theory and a review of the Knox or van Deemter equations. While band broadening is a major driver in the observed speed and resolution, other areas also contribute to the benefits of the method. I will discuss two: system volume and the geometry of the particles and columns.
For those who have tried to implement fast LC and found poorer resolution, there can be a number of reasons. Following is one anecdote that I have heard more than once. A scientist related to me that he had purchased a 50 mm × 2.1 mm column packed with sub-2-µm C18 packing material. To run solvent through the column, the flow rate had to be below 0.5 mL/min. The resulting peaks were broad and the back pressure was a little over 300 bar. There were two factors working against this chromatographer's separation. The low flow rate contributed to a larger plate height, and the system volume was not minimized. These causes are explained below, under system volume and theory.
System Volume
In designing the current UHPLC systems, the engineers readily understood that one of the important considerations was to minimize the internal volume ahead of the column.
UHPLC systems typically have internal volumes of 125–270 µL. Conventional HPLC systems typically have internal volumes of 600–1200 µL. This is very important in gradient formation. With less system volume, the gradient is formed efficiently and the eluent change gets to the head of the column faster.
With conventional HPLC systems, there are modifications that the HPLC manufacturers suggest to make the system more compatible with fast LC. Much of this work is aimed at reducing the internal volume ahead of the column. The internal volume can be reduced by 30–50% to achieve faster gradient formation. However, with fast LC, one can find that the higher flow rates, frequently 3–5 times the internal volume, also can aid in minimizing the effect of the internal volume on the gradient, which can yield higher resolution.
Smaller detector cells can aid in enhancing resolution as well. All of the column connections need to use 0.005-in. tubing to minimize peak broadening after the column to the detector.
Theory
Returning to the van Deemter equation, it is clear that faster flow rates can yield a smaller plate height because of the decrease in the "B" term with the higher mobile phase velocity. The decrease in particle size is significant for decreasing the "A" term. The smaller particle size also affects the "df" term in that with a smaller particle, the diffusion will be faster since there is less distance to travel in the particle:
H: height equivalent to a theoretical plate (HETP)
u: mobile phase velocity–flow rate
dp: particle diameter
k: retention factor
DL and DS: solute diffusion coefficients for mobile and stationary phases
λ: constant related to particle geometry
γ: factor related to interstitial volume and flow resistance
df: stationary film thickness
However, with smaller particles and faster flow rates, there is higher back pressure.
Geometry
As we consider the geometry of the column and particle size, we start to understand some of the other elements that contribute to the resolution and speed observed with fast LC.
The data in Table I show that the surface area of a 5-µm particle is almost eight times larger than a 1.8-µm particle, but 22 1.8-µm particles can fit in the volume of a 5-µm particle. Since the actual packing efficiency of spheres is about 68%, the real number of particles is 14.6. The effective volume is the number of 1.8-µm particles that could "fit" inside the volume of a 5-µm particle. Thus, the effective surface area of the smaller particle packing is nearly twice that of the 5-µm packing. This is one more way that smaller particles can yield higher resolution or faster analyses. In a column volume packed with 1.8-µm packing, the surface area is twice that of a 5-µm material.
Table I: Particle size dimensions
Table II shows that a 2 cm × 0.46 cm column packed with 1.8-µm packing material has a surface area equivalent to a 5 cm × 0.46 cm, 5-µm column, whereas the 5 cm × 0.21 cm column is only about one-quarter of the surface area.
Table II: Column dimensions
Although I am discussing the effective surface area of the particle, what we really are looking at is the carbon load. However, if the column chemistries are the same, then this discussion is still applicable.
Applications
I will now review a couple of applications where fast LC yielded better results over the traditional HPLC methods.
Our company embarked on a program where we would synthesize more than 200,000 compounds. The libraries of compounds needed to be assayed as soon as possible after synthesis. We used combinatorial synthesis on solid support to generate the library compounds. After they were synthesized, the compounds were cleaved from the support, arrayed into 2-mL 96-well mother plates and formatted into 384-well plates for a quality assessment using HPLC with diode-array and mass spectrometry (MS) detection.
The libraries varied in size from 6000 to 28,000 compounds, each needing a determination of purity using UV at 210 nm and MS to confirm the correct mass of the compound.
We had four HPLC–MS systems and needed to complete the QC of the plates within 1–2 weeks of receiving a library. We had been using an HPLC method employing a 50 mm × 20 mm, 5-µm packed column, which had a gradient time of 4 min and an overall cycle time of 5.25 min. The method was robust for what we were doing and seemed adequate to that point. However, at 11 samples/h, we would only be able to run 1000 samples/day. I began to consider how we could run samples faster and not lose resolution.
UHPLC systems had been introduced but I was concerned that, as with many first generation systems, it might not be robust. Therefore, we continued to use traditional HPLC systems. I wanted to evaluate the new 1.8-µm packed columns, yet as I considered the back pressure that we already had with 50 mm × 2 mm, 5-µm packed columns, I did not see how to run columns packed with 1.8-µm particles with our existing HPLC systems.
I recognized that higher flow rates could yield faster analyses, and smaller particle sizes could yield higher resolution, but the back pressure would be very high. At this point, I realized that changing from a 50 mm × 2.1 mm column with 5-µm packing to a 20 mm × 4.6 mm column with 1.8-µm packing would be doable. The dimensions of the change in column radius and particle size almost cancel out. This would yield a column that could run at a higher flow rate with similar back pressure and potentially higher resolution.
Table III: Flow rate and back pressure optimized for di-octyl phthalate (DOP)
A mixture of uracil, hydrocortisone, reserpine, and di-octyl phthalate (DOP) was used to develop the library separation. These compounds represent a log P range of –2 to 9. To see how fast the separation could be performed, DOP was used for optimizing the time because this was the last eluted compound. The data in Table III show the impact of flow rate on retention time and back pressure and the resulting gradient retention factor k* and resolution. The flow rate and back pressure had to be balanced to yield short retention times and reasonable Rs and k*. The resolution was flat through the series of flow rates. The shortest retention time would be best, but whether I used 2.5 or 3.0 mL/min flow rates, there was not a significant change in time and the back pressures, Rs, and k* were acceptable. Although the 4.0-mL/min flow rate has a shorter retention time, the back pressure is higher and solvent consumption is significant. These are important considerations when running thousands of compounds, where back pressure may increase over time, and systems run over nights and weekends unattended.
Table IV: Optimization of flow rate based on the k* of hydrocortisone, reserpine, and di-octyl phthalate (DOP)
Table IV shows that k* is flat in response for 2.5 mL/min and 3.0 mL/min. Therefore, the lower flow rate was used, which yielded the final 2.5 min chromatography observed in Figure 1.
Figure 1: Chromatograms from two methods separating uracil, hydrocortisone, reserpine, and di-octyl phthalate (DOP): 4-min gradient separations on 5 cm à 0.21 cm, 5-µm packing (top); 2.5-min gradient separation on 2 cm à 0.46 cm, 1.8-µm packing (bottom).
To see the effect of the smaller particles on retention and peak shape, see Figure 1. Note that the observed peaks in the smaller particle column are sharper and have higher resolution and k* than the 5-µm packed column.
Table V shows the metrics for the 4-min method that had been used in the laboratory versus the 2.5-min method. The k* and resolution demonstrate the higher efficiency of the faster method. Because the purpose of this method was to determine the purity in the UV and determine whether the expected molecular ion was present, it was important to have as high a reasonable resolution as possible across the entire gradient.
Table V: Comparison of HPLC method metrics for the 2.5-min assay and 4-min assay
A library compound (see Figure 2) was run on both methods. The 0.48-min peak in the 4-min method is resolved at 0.65 min in the 2.5-min method. The sharper resolution aided quality assessment of the library and allowed 2000 samples a day to be run with four LC–MS systems, yielding faster analysis and better resolution.
Figure 2: Chromatograms of a library compound run on both methods.
To demonstrate the robustness of smaller particles, Figure 3 shows the same column where the standard solution of uracil, hydrocortisone, reserpine, and DOP with the initial run and then after 2500 injections. No loss of resolution, or significant back pressure increase, was observed.
Figure 3: Standard run on columns initial and after 2500 injections to show particle/column robustness.
One last application will address the practical concern of quantitation using a fast LC method for pharmacological dose verification.
In the course of one of our pharmaceutical development programs, we needed to supply doses of our drug for our pharmacology group. After dose preparation, samples had to be tested to ensure the proper dose had been prepared. To assay the dose, an HPLC method was used that took 34 min to run. At that juncture, doses were prepared in the morning for early dosing, which meant that the dosing by pharmacology was at risk until the dose verification was confirmed. With a blank standard and the dose itself, it could be 3 h until the dose was verified. A faster HPLC method was required.
The following method was used for dose verification, and was converted to a fast method. The chromatography is displayed in Figure 4.
Figure 4: 20 scans across 1.8-s wide DFTPP peak showing expected resolution.
34-min Method: Column: 150 mm × 4.6 mm Symmetry; mobile phase A; water with 0.1% trifluoroacetic acid; mobile phase B: acetonitrile with 0.1% trifluoroacetic acid; flow rate: 1.0 mL/min; gradient:
The 13.1-min impurity ahead of the 13.8-min drug was a critical separation that was necessary to maintain to quantitate drug in the dose solution.
The developed separation yielded an acceptable separation and afforded a cycle time of 9 min, which allowed completion of the dose verification in 1 h.
Following is the method used for the fast LC dose verification. The chromatography is shown in Figure 5.
Figure 5: 9-min fast method for dose verification.
9-min Method: Column: 50 mm × 4.6 mm Agilent SB C18; mobile phase A: water with 0.1% trifluoroacetic acid; mobile phase B: acetonitrile with 0.1% trifluoroacetic acid; flow rate: 1.5 mL/min; gradient:
The statistical analysis for the dose verification results are displayed in Table VI. The standard deviation of the fast method is significantly reduced when compared with the original method. Thus, a fast LC method was developed which reduced the analysis time by 74%, and provided lower variability.
Table VI: Comparison of quantitation: fast LC (9 min) vs. the 34-min method
These data demonstrate that a faster method can yield reduced variability without losing the ability to separate closely eluting impurities near a product peak.
In conclusion, the main reasons for using fast LC have been highlighted in this paper: productivity and better resolution. With a reasonable understanding of the HPLC system volume and ways to reduce it, a recognition of how particle size and flow rate affect band broadening, and the geometry of particles, much faster methods can be developed readily. Concerns by some chromatographers no longer have merit. Fast methods do not have to yield lower resolution; in fact, the resolution can be higher. Columns do not have to suffer shorter life times, but can yield as many injections as those used in traditional methods. Reproducible quantitation of closely eluting peaks in fast LC is achievable because separations can be very robust.
The issue of methods used in a GMP environment being costly to revalidate is a legitimate reason to keep a slower methods. Otherwise, new method development efforts should be aimed at using fast LC methods wherever possible. In less regulated environments, it is hard to justify not employing fast LC on a routine basis.
Joseph Helble is Senior Scientist with Infinity Pharmaceuticals, Inc., Cambridge, Massachusetts.
References
(1) M.W. Dong and J.R. Gant, LCGC 2, 294 (1984).
(2) J.J. van Deemter, F.J. Zuiderweg, and A. Klinkenberg, Chem. Eng. Sci. 5, 271–289 (1956).
Articles in this issue
Newsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.
Advertisement
Related Content
Advertisement


Advertisement
Advertisement
Trending on LCGC International
1
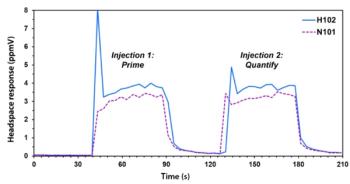
High-throughput Headspace Analysis of Volatile Nitrosamines and their Secondary Amine Precursors
2
Streamlined Method Development for Efficient and Reliable Lipid Nanoparticle Analysis
3
Best of the Week: The Scientific Method in the Age of AI, Rewiring the Fundamentals
4
Detecting Estrogen Signatures in MCN Tumors via Targeted LC-MS/MS
5
