Utilizing Multidimensional LC–MS for Hydroxyl Radical Footprinting Analysis
Special Issues
The potential of multidimensional online peptide mapping analysis as a strategy for improving a postlabeling workflow for protein–protein interactions is demonstrated using both hydroxy radical footprinting–mass spectrometry (HRF–MS) and LC–MS/MS.
Research tools that can decipher protein–protein interactions and binding interface contact points can aid in the successful development of biotherapeutics. Hydroxyl radical footprinting–mass spectrometry (HRF-MS) technologies are being developed as tools for deciphering protein–protein interactions. We have demonstrated the utility of using fast photochemical oxidation of proteins (FPOP) as an HRF-MS technology for biotherapeutic protein structural characterization and analysis of protein–protein interfaces; monoclonal antibody (mAb) epitope mapping has also been demonstrated using this technique. However, the postlabeling workflow that utilizes offline protein digestion and liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis is labor intensive and time consuming, and has significant sample consumption. In the present work, we demonstrate the potential of multidimensional online peptide mapping analysis (reduction, tryptic digestion, and separation) as a strategy for improving our postlabeling workflow for protein–protein interactions. The proof-of-concept studies were performed using a Fab antibody fragment and its antigen binding partner.
Monoclonal antibody (mAb) structural characterization and their epitope mapping are both important aspects of biotherapeutic discovery to further understand structure-function relationships, mechanisms of action, and antibody–antigen binding interactions (1). The most commonly used techniques for epitope mapping are nuclear magnetic resonance (NMR) and X-ray crystallography, which provide high-resolution information on binding interactions at the atomic level (2,3). However, these techniques may have limited biological relevance, most notably for the analysis of structure and dynamics of proteins in conformation states of interest, and are especially challenging for large, complex proteins. On the other hand, spectroscopy-based approaches such as Fourier transform–infrared spectroscopy (FT-IR) (4), analytical ultracentrifugation (AUC) (5), and fluorescence spectroscopy (6) can be used for mAb structural characterization, providing residue-specific information or high-throughput analysis, or both. However, these methods are not suitable for localizing specific areas or regions of structural change in the proteins, and are generally considered as low resolution techniques/
Alternatively, footprinting based on liquid chromatography–mass spectrometry (LC–MS) has recently emerged as a powerful approach for the structural characterization and elucidation of protein–protein interactions (7,8). These techniques are considered bottom-up MS approaches to protein analysis in which proteins are labeled, subsequently digested, and then analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS), providing information on solvent accessibility for specific chains in the protein structure. Information on the solvent accessibility of the peptides can be encoded onto the peptides by means of either reversible (hydrogen/deuterium exchange [HDX]) or irreversible (hydroxyl radicals) chemical probes (1).
Another growing technology in the epitope mapping field is hydroxyl radical footprinting–mass spectrometry (HRF-MS) (9–11). HRF-MS uses a high powered laser to photodissociate hydrogen peroxide into hydroxyl radicals, enabling the covalent and irreversible labeling of protein residues. The addition of hydroxyl groups to protein side chains induces a +16 mass shift, easily detectable by tandem MS analysis. Because of the irreversible labeling reaction, no extra care is needed for the postlabeling workflow. However, the conventional approach for the postlabeling workflow involves time-consuming offline procedures, with reduction and alkylation of the protein and buffer exchange, followed by an offline tryptic digestion before injection onto an LC–MS instrument. In addition, the tryptic digestion in solution also suffers from a number of drawbacks such as low efficiency and auto-digestion of the enzyme (12).
Recently, fast, online, and automated multidimensional LC workflows have been developed on commercial ultrahigh-pressure liquid chromatography (UHPLC) systems for the reduction and digestion of proteins, bypassing the offline manual sample pretreatment reduction and digestion of mAbs (13). The use of these instruments for automated and fast digestion under high pressure with immobilized trypsin cartridges significantly reduces the digestion times compared to the standard offline approach (minutes vs. hours). Using the multidimensional LC–MS approach (automated online reduction, digestion, and separation) for the postlabeling HRF-MS workflow can significantly reduce sample handling and operator time compared to standard offline procedures. In this study, we explore this possibility of using a multidimensional LC workflow to analyze the known paratope (the complementarity determining region, [CDR]) map of a monoclonal Fab fragment against its antigen, and compare the results against the conventional offline procedure.
Experimental Method
Reagents and Sample Preparation
Millipore Sigma provided the hydrogen peroxide (30%), dithiothreitol (DTT), sodium iodoacetate (IAA), and all MS-grade solvents (2-propanol and acetonitrile). Tris(hydroxymethyl) aminomethane (TRIS) base, potassium phosphate monobasic, potassium phosphate dibasic, potassium chloride, MS-grade formic acid (FA) and trifluoroacetic acid (TFA) were all purchased from Sigma-Aldrich. Calcium chloride dihydrate and sodium chloride were obtained from BeanTown Chemicals. Water was obtained from a Milli-Q purification system from Millipore. MS-grade water, 0.1% FA in water, 0.1% FA in acetonitrile and tris(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) were all purchased from Thermo Fisher Scientific.
The antigen and the Fab were produced in Escherichia coli cells and prepared for labeling by size-exclusion chromatography. Samples were purified and buffer exchanged using a TSKgel UP-SW3000 column (4.6-mm I.D. x 30 cm, 2-µm) purchased from Tosoh Bioscience and a 0.2 M potassium phosphate, 0.25 M potassium chloride, pH 6.2 buffer. Samples were collected off the column and diluted to approximately 2 mg/mL for labeling. The antigen–Fab complex was created by mixing the components at a 1:2 antigen–Fab ratio.
Hydroxyl Radical Labeling Procedure
Samples were subjected to labeling as previously described (14). Briefly, a high power laser was focused on a glass capillary used to irradiate the sample. Two syringes, one containing the protein samples (Fab or antigen–Fab complex) combined with arginine (used to help control the amount of hydroxyl labeling), and the other syringe (containing hydrogen peroxide), converged on a T-mixer just before the point of laser irradiation. This T-mixer allowed the solution to mix at a 2:1 (protein:hydrogen peroxide) volume ratio. After irradiation, the samples are delivered in-line to a sample tube containing methionine and catalase to quench the labeling reaction.
Standard Off-line Peptide Mapping Approach
Hydroxyl radical labeled protein samples were digested as previously described (14). Briefly, the proteins were reduced with guanidine HCl (6 M) and DTT (10 mM, 45 °C for 10 min), and alkylated with IAA (25 mM, at room temperature for 5 min), and quenched with DTT (50 mM, room temperature). The resulting samples were then desalted using NAP-5 columns (GE Healthcare), digested with trypsin (5 μg, 37 °C, overnight), and quenched with 100% of FA.
Tryptic peptides (10 μg) separation was performed using a Waters H-Class UPLC system with a Waters Acquity UPLC CSH130 C-18 column (1.7-μm, 2.1 mm × 150 mm). The flow rate was fixed at 0.3 mL/min and the column temperature at 77 °C. Mobile phases consisted of 0.1% FA in water (A) and acetonitrile (B). The gradient conditions consisted of 0 to 35% B in 42 min. The MS analysis was carried out with a ThermoFisher QExactive Orbitrap MS operating in positive mode, performing MS2 scans on the top-10 most abundant peaks in data-dependent acquisition mode in the m/z range 350–2000 at a resolving power of 35,000. MS data treatment was performed using Byonic and Byologic Footprint Software Suites (Protein Metrics, Inc.) for peak identification and quantitation of percent modification for each peptide, respectively. All samples were analyzed in triplicate.
On-line Peptide Mapping Analysis
Instrument
All online peptide mapping analyses were carried out on a biocompatible ThermoScientific Dionex UltiMate 3000 Rapid Separation (RS) system. It is composed of two RS dual pump modules, each containing two ternary pumps, two thermostatic RS column compartments, and an RS auto sampler. The multidimensional LC setup was coupled to a Q Exactive Orbitrap MS. For multidimensional LC–MS analysis, the same MS parameters for the standard offline peptide mapping approach were used.
1st Dimension (â1D): On-column Reduction
Hydroxyl radical labeled proteins were first loaded and reduced on column using the BioResolve RP mAb Polyphenyl column (2.1-mm I.D. x 50 mm, 2.7-µm, 450 Å) purchased from Waters. Mobile phases A and B contained 0.1% FA and 0.05% TFA in water and acetonitrile, respectively. Mobile phase C contained the TCEP reducing agent at 25 mM in acidified water (pH 4.5). The valve located between the second- and third-dimension columns was switched at 20 min when the elution starts to divert the reduced proteins to the third dimension trypsin cartridge. Supplemental Table S1 lists the gradient and valve details.
2nd Dimension (â2D): Tryptic Digestion in Flow-Through Mode
The Poroszyme immobilized trypsin cartridge (2.1-mm I.D. x 30 mm), supplied by Thermo Fisher, was equilibrated from 0 to 20 min, then set in-line with the 1D and 3D reversed-phase LC columns from 20 to 50 min, for the online digestion and subsequent trapping of the peptides on the 3D reversed-phase LC column. Digestion buffer was composed of 10 mM calcium chloride and 50 mM TRIS pH 8.0, whereas a 2-propanol/acetonitrile (70:30, v/v) mixture was used to wash the trypsin cartridge after each cycle.
3rd Dimension (ââ3D): Peptide Mapping Analysis
The trapped peptides from 20 to 50 min were then eluted between 50 min and 100 min from an XSelect CSH C18 column (2.1-mm I.D. x 150 mm, 3.5-µm, 450 Å) supplied by Waters. MS grade mobile phases A and B were composed of 0.1% FA in water and acetonitrile, respectively. Table S1 lists the gradient and valve details.
Results and Discussion
Off-line Versus On-line Workflows
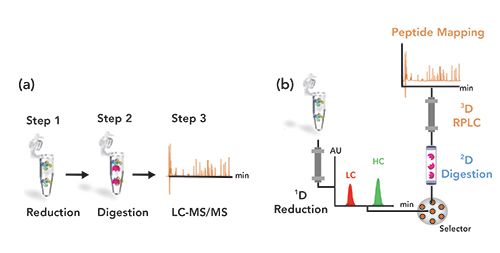
Figure 1 shows the two workflows for the (a) conventional offline and (b) online peptide mapping approaches. With the online peptide mapping workflow, the Fab sample was injected onto the 1D reversed-phase LC column, where it was subsequently trapped at the head of the column. For the reduction of the Fab into reduced subunits (heavy chain and light chain), a 25 mM TCEP solution in acidified water (pH 4.5) was used. The reduced Fab chains eluted from the 2D column and were then digested online on the immobilized trypsin cartridge, prior to their separation on the 3D reversed-phase LC coupled to MS.


Figure 1: Schematic representation of a) conventional offline and (b) online peptide mapping (reduction-reversed-phase LC x tryptic-digestion x reversed-phase LC–MS) workflows for hydroxyl radical footprinting analysis.
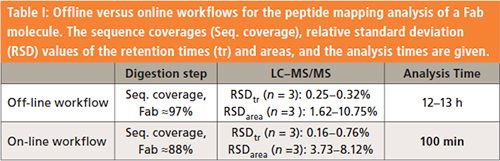
The precision of the online multidimensional LC digestion workflow was compared to the standard offline procedure to assess whether there were differences between the workflows and whether the multidimensional LC approach could be used for hydroxyl radical footprinting analysis. A Fab molecule was selected and three consecutive injections were performed on the online system and compared with a conventional offline procedure (Table I). A sequence coverage of 88% using the online workflow, compared to the offline value of 97%, was achieved, and the sequence coverage remained consistent for each experiment. The difference in sequence coverage results from the absence of a single peptide with the online approach, which is discussed in the next section. The repeatability of the online peptide mapping method was then assessed by performing triplicate analysis. For this purpose, four unmodified peptides in common with the offline approach were selected and compared according to their retention times and peak areas. As shown in Table I, the results demonstrated a repeatability of the online analysis with relative standard deviation (RSD) values lower than 0.8% and 8.2% for the retention times and peaks areas, respectively.
The leading advantage of the multidimensional LC online peptide mapping approach is the short analysis time with a turnaround of 100 min compared to the conventional offline procedure, which typically requires several hours (Table I). The decrease in analysis time is achieved by the automation of all the labor-intensive and manual sample preparation steps associated with the conventional offline approach, including reduction/alkylation, desalting, digestion, and buffer exchange. Another advantage of the online approach is the low amount of sample required (10 µg per injection for the online peptide mapping analysis, compared to 100 µg typically required for the offline digestion), resulting from the low efficiency of the tryptic digestion in solution.



Off-line versus On-line HRF-MS Results
The amount of oxidation that occurs during HRF labeling for a given region of a protein is proportional to the solvent accessibility of the region. Comparing the oxidation of Fab peptides in the unbound and bound antigen–Fab complex state allows for paratope mapping of the antigen–Fab interaction, where the regions showing a decrease in oxidation in the bound state are possible binding paratopes. The CDR-containing peptides, the known paratopes in this interaction, were analyzed for both offline and online digested samples to understand if the online digestion method could be used for the paratope mapping analysis from HRF labeling.
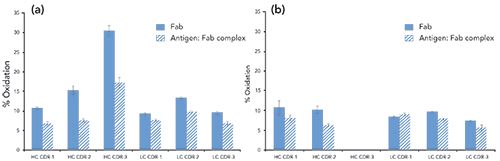
Data analysis was conducted similarly between the offline and online samples (see experimental section). The offline peptide mapping samples indicated a decrease in oxidation (possible binding paratope) in the antigen–Fab complex sample compared to the Fab sample in peptides containing the CDRs (Figure 2). The multidimensional LC online peptide mapping results also indicated a decrease in oxidation in most of the CDR peptides detected with the offline procedure (4 out of 6 CDR-containing peptides), showing confidence in the use of the online automated approach for the HRF-MS paratope mapping workflow. Some of the differences between the online and offline results include the heavy chain CDR 3 peptide not being detected by the online digestion procedure, and the light chain CDR 1 peptide showing no significant difference in oxidation between Fab and antigen–Fab complex after online digestion. These differences are most likely attributable to the elution gradients in multidimensional LC–MS, where the very hydrophobic heavy chain CDR 3 peptide is retained on the 3D reversed-phase LC column. Further method optimization of the online workflow is needed to increase the sequence coverage and recover the CDR 3 peptide. However, overall this proof of concept study shows that there is general agreement between the results generated with the offline and online digestion procedures, showing that HRF-MS analysis can be performed using the multidimensional LC online digestion procedure to make data acquisition faster and easier.
Figure 2: Hydroxyl radical footprinting results for unbound Fab and antigen:Fab complex for (a) offline HRS-MS procedure, and (b) online HRS-MS procedure. HRF analysis was completed on the Fab heavy and light chain CDR-containing peptides.
Conclusion
In this work, we introduced an innovative multidimensional LC–MS setup for a fast (100 min per run), automated, and online peptide mapping workflow for postlabeling HRF-MS epitope mapping analysis. Although the heavy chain CDR 3 peptide was not detected by the online approach, paratope mapping results were generally similar between the online and offline workflows. From these results, we can conclude that with further optimization, the online multidimensional LC system can be used for hydroxyl radical footprinting analysis to reduce the postlabeling workflow time from >10 h to 100 min.
Acknowledgments
The authors acknowledge Matt Kalo and Jennifer Rea from Genentech (South San Francisco, California, USA) for their support.
Supplemental Information
A supplemental table, showing on-column reduction and subsequent separation of reduced proteins, followed by on-line tryptic digestion and peptide mapping analysis, can be found online in the issue archive where this article appears.
References
- K. F. M. Opuni et al., Mass Spectrom. Rev. 37(2), 229–241 (2018).
- O. Carugo and P. Argos, Protein Sci.6(10), 2261–2263 (2008).
- Q. Xu et al., J. Mol. Biol.381(2), 487–507 (2008).
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin.39(8), 549–559 (2007).
- J. T. Pelton and L. R. McLean, Anal. Biochem.277(2), 167–176 (2000).
- O. Tcherkasskaya, V. E. Bychkova, V. N. Uversky, and A. M. Gronenborn, J. Biol. Chem.275(46), 36285–36294 (2000).
- A. Artigues et al., in Modern Proteomics–Sample Preparation, Analysis, and Practical Applications, H. Mirzaei and M. Carrasco, Eds. (Springer International Publishing, Cham, 2016). pp. 919, 397–431.
- H. Zhang, W. Cui, and M. L. Gross, FEBS Lett.588(2), 308–317 (2014).
- A. T. Wecksler, M. S. Kalo, and G. Deperalta, J. Am. Soc. Mass Spectrom.26(12), 2077–2080 (2015).
- G. Deperalta et al., mAbs5(1), 86–101 (2013).
- J.G. Kiselar and M.R. Chance, J. Mass Spectrom. 45(12), 1373–1382 (2010).
- S. Perchepied et al., Talanta206, 120171 (2020).
- C. Gstöttner et al., Anal. Chem.90(3), 2119–2125 (2018).
- M. Lin, D. Krawitz, M. D. Callahan, G. Deperalta, and A. T. Wecksler, J. Am. Soc. Mass Spectrom.29(5), 961–971 (2018).
Julien Camperi, Arthur John Schick, and Aaron T. Wecksler are with the Protein Analytical Chemistry group at Genentech Inc., in South San Francisco, California. Davy Guillarme is with the School of Pharmaceutical Sciences at the University of Geneva, in Switzerland. Direct correspondence to: wecksler.aaron@gene.com













, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

