Considerations and Advances in Developing Analytical Assays for Measuring Product-Related Variants in Bispecific Antibodies
Given the complexity of producing bispecific antibodies, suitable analytical methods to detect and measure the levels of undesired variants are essential. Here, a novel MS-based method for variant detection is presented.
Bispecific antibodies open new opportunities in drug development and have become a desirable drug format in cancer immunotherapy. In this type of application, one arm of the bispecific antibody binds to a cancer-specific receptor, while the other arm binds to a T-cell specific receptor to induce cancer cell elimination. Because up to four different polypeptide chains are involved in the assembly of a full-length bispecific antibody, many strategies have been developed to maximize the yield of the desired product (heterodimer) and facilitate the removal of undesired variants, such as homodimers. The successful adaptation of each strategy requires the development of suitable analytical methods to detect and measure the levels of undesired variants. Because the physicochemical properties of the undesired variants can be very similar to those of the desired heterodimer, it is paramount to magnify the minor differences as part of method development. In addition, given that the facilitation of heterodimerization of bispecific antibodies often relies on specific mutations that may impact the physicochemical properties, additional method assessments may need to be considered to ensure their suitability, such as interference from desired product to undesired variants. In the first half of this article, a summary of the common strategies employed in the production of full-length bispecific antibodies is provided. Then two case studies are presented in the second half. In the first case study, the proof-of-concept results are shared for a novel analytical method for variant detection using hinge-specific proteases that cleave at defined sites above or below the disulfide bonds in the hinge region. In the second case study, the method qualification experiments are described for a homodimer quantitation method to address the potential charge–charge interaction between a homodimer and the desired heterodimer.
Bispecific molecules are molecules that can simultaneously bind to two targets with high specificity, and are now emerging as a growing class of cancer immunotherapies (1–3). They include full-length bispecific antibodies, Fc-less bispecific molecules, fusion proteins, and many other forms of engineered proteins (3). Bispecific antibodies have many desired properties that are made possible by simultaneous binding to two targets. This class of therapeutic proteins (4) has been applied or is being tested for a wide range of applications, such as 1) inducing cancer cell killing by T-cells or natural killer (NK) cells through simultaneous binding to a cancer cell surface marker and an immune cell surface antigen, such as CD3; 2) activating or inhibiting a specific cellular pathway through simultaneous binding to two cell-surface targets; and 3) bridging IX and X factors in the coagulation cascade for the treatment of hemophilia A (1,3,5).
The concept of bispecific antibodies dates back to the last century (6,7). However, clinical success and large-scale production only became a reality in the past few years (2), coinciding with growing interest in the vast potential of this class of molecules among both the medical and scientific communities (3). That passionate interest spurred tremendous technical advancements in the development, engineering, and production of bispecific molecules. This article focuses on full-length bispecific antibodies and the analytical strategies required to ensure a successful product outcome. An understanding of the potential variants and manufacturing options to reduce their presence is also discussed. In general, there are two strategies to maximize the yield and purity of the desired product: to encourage heterodimerization (increasing the level of desired product), or to facilitate homodimer removal (8).
Strategies to Encourage Correct Chain Pairing
To achieve high yield and purity of the desired product when producing a bispecific antibody, the most effective strategy is to minimize the generation of undesired product variants by encouraging correct chain pairing (8). For a full-length bispecific antibody, there are typically four different polypeptide chains, light chain 1 (LC1), light chain 2 (LC2), heavy chain 1 (HC1), and heavy chain 2 (HC2). When all four polypeptide chains are produced from a single cell, each chain has an equal opportunity to form disulfide bonds with the others. In such a case, one could theoretically generate 10 different products with only one of them being the desired product (1/10) (Scheme 1). One strategy to simplify manufacturing is to reduce the number of possible chains, such as using the common LC or common HC. By reducing the total number of different polypeptide chains to three (common LC, HC1, and HC2; or LC1, LC2 and common HC), the theoretical number of different products reduces to only three. Such a strategy can significantly reduce the possible number of undesired products from nine to two, but may not always be possible due to other considerations, such as desired affinity to target or patent-related considerations.


Scheme 1: Potential variants produced during the assembly of a full-length bispecific antibody. LC = light chain. HC = heavy chain.
Another strategy to encourage the formation of the desired product is achieved by introducing specifically designed mutations in the CH3 domain of the Fc or hinge region of the HCs; these mutations introduce additional features in each HC to encourage heterodimerization and discourage homodimerization. Although the theoretical number of different products in this case is still 10 (9 of which are undesired), the likelihood of forming the desired product is significantly increased. To date, more than a dozen different mutations have been engineered and demonstrated as effective in maximizing the yield of the desired product (9–16). These features include mutations with increased side chain size and hydrophobicity (“knobs”) in one HC, matched with another HC with mutations that form hydrophobic “holes” (10). These mutations encourage the desired HC pairing through hydrophobic interactions and discourage the formation of knob–knob homodimers by steric hindrance. This strategy has been used in both single-cell format or two-cell format. For the two-cell format, the two halves of the antibody (LC1 + HC1 and LC2 + HC2) are produced separately in two different cell lines before annealing under a mild in vitro reduction condition (17).
Another example of Fc mutation-driven chain pairing involves the introduction of amino acid mutations containing charged side chains that can form salt bridges between two different HCs (15). Similarly, desired chain pairing between the HC and LC can be facilitated using this strategy (18).
An additional strategy specifically aimed at reducing LC mispairing is the “CrossMab” format, where the HC and LC domains in Fab are swapped (19). This domain swap maintains antigen-binding affinity while reducing the possibility of LC mispairing by making the two LCs very different. The main advantage of this strategy is that it can be applied to any two existing full-length antibodies to generate bispecific antibodies without screening for new complementarity-determining region (CDR) sequences.
Currently, these strategies have been employed in several clinical-stage bispecific antibodies.
Strategies to Facilitate the Removal of Homodimers
The undesired products are removed through the purification process as much as possible. If certain forms of the undesired products share many similar characteristics with the desired product, the robust removal of these undesired products during the purification process becomes a challenge. Therefore, it can be ideal to engineer the polypeptide chains in a bispecific molecule to create distinct characteristics, such as hydrophobicity and isoelectric point (pI), so that the undesired products can be more easily separated from the desired product (8).
The purification process of full-length antibodies typically involves three columns (10,11,20). The first is a protein A affinity capture column to extract the antibodies from host cell proteins (HCPs) and other cell culture impurities; noting that full-length antibodies bind to protein A via the Fc region with high specificity. The second and the third columns are called “polishing” columns; their purpose is to further remove HCPs and product-related variants, such as aggregates and fragments. These columns are typically ion-exchange or mixed-mode chromatography columns.
The first affinity capture column can be used to differentiate heterodimer products from the homodimer variants (11). Mutations in the protein A binding region of the Fc domain can be introduced to modulate the binding affinity of bispecific antibody products to the protein A column. As a result, the homodimers have either strong or weak affinity to the protein A column, while the binding affinity of the heterodimer is at an intermediate level.
The latter columns in the purification process have also been exploited to separate desired product from the homodimers (9). One interesting technique introduces mutations in the CH3 domain of both HCs, so that the pI values of both homodimers are significantly different from that of the heterodimer. The different pI values allow for the complete removal of homodimers through the commonly used ion-exchange column in the purification process. The different pI values are achieved by introducing mutations with charged side-chains (such as lysine or aspartic acid) so that the homodimers bear a different net charge from either homodimer. In large-scale purification processes where bind-and-elute mode is often used, the net charge difference between the desired product and the homodimers should be large enough to allow sufficient removal of the homodimers. When the complete removal of both homodimers by one column step is not possible, other factors, such as the expected safety impact of the homodimers, needs to be considered to achieve optimum purification conditions. For example, if one homodimer is assumed to possess a higher safety risk than the other homodimer, the removal of the former should be prioritized. Meanwhile, the assessment of safety and efficacy impact of both homodimers should be continued as the drug candidate progresses through clinical phases.
Analytical Challenges to Identify and Quantitate Product-Related Variants
Even with advanced manufacturing and purification techniques, identification and quantitation of undesired product variants remains a necessity but can be a difficult challenge for a bispecific antibody, especially in cases where mutations have been introduced (21–23). Mass spectrometry (MS) has been the most capable tool for identifying and quantitating these variants, especially during the early stages of development (23–27). For methods developed to monitor product variants, purified analytes, such as homodimers, are usually required as standards to obtain quantitative results through a standard curve generated by serial dilution or standard addition. Whenever possible, a low quantitation limit and wide dynamic range have been demonstrated using these approaches.
As depicted in Scheme 1, the product-related variants in bispecific antibodies include homodimers and the undesired chain-pairing variants (mismatched LC, common LC, and other chain-pairing variants). The undesired chain-pairing variants can be very challenging to detect and quantitate for several reasons. They are usually present at much lower levels relative to the desired product, and may present in a mass region crowded by the expected variants (such as post-translational modifications or PTMs) of the desired product. In such a case, an orthogonal approach such as MS analysis of the subunits is valuable to confirm the presence of the undesired chain-pairing variants. The LC mismatched species (LC1HC2 + LC2HC1) have the added challenge of being structural isomers to the desired product, and cannot be distinguished at the intact or reduced level by MS.
Recent methods that employ hinge-specific proteases (such as IgdE, Kgp, or IdeS) that allow subunit analysis by MS have demonstrated strong potential for the detection and potential quantitation of LC chain-pairing variants (22,24) and a proof-of-concept example will be presented later.
In addition to MS-based methods, chromatography and capillary electrophoresis (CE)-based methods are also widely used for the detection and quantitation of product variants. In the second case study presented here, the homodimers have pI values different from that of the heterodimer, due to the mutations that were introduced to facilitate the removal of homodimers by ion-exchange chromatography. However, the heterodimer is presumed to contain a positive charge patch, thus it is important to consider potential charge-to-charge interactions between the heterodimer and the negatively charged homodimer (homodimer 1). In this case study, we presented the experiments to confirm the linearity of homodimer 1 quantitation that supported a successful qualification of this chromatography-based method for homodimer quantitation.
Case Studies
Detection of Chain-Pairing Variants in Bispecific Antibody A by MS Analysis of Subunits
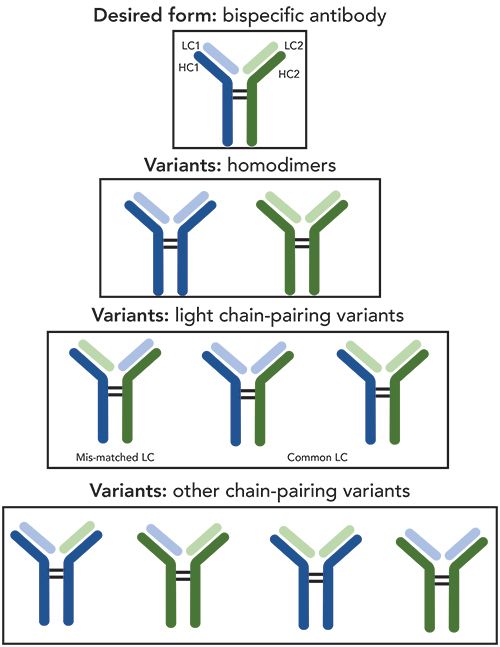
As discussed in the previous section of this article, the mismatched LC variants and the common LC variants (Scheme 1) can pose a significant challenge in analytical method development. To detect these LC-pairing variants, MS-based analytical assays for bispecific antibody A that involve two different hinge-specific enzymes were developed. As depicted in Scheme 2, the desired product (heterodimer) is isobaric to the mismatched LC variant at both intact and reduced levels.

Scheme 2: Strategies for chain-paring variant detection using hinge-specific enzymes.
Upon enzyme treatment, a ~100 kDa subunit of F(ab)′2 containing both LCs and two HC fragments (Fd′) is generated. Given that Fd′ fragments usually bear the majority of the common PTMs, such as N-glycosylation, glycation, and oxidation, the MS spectra in the mass range of the F(ab)′2 subunits can be significantly simplified, allowing the sensitive detection of common LC variants.
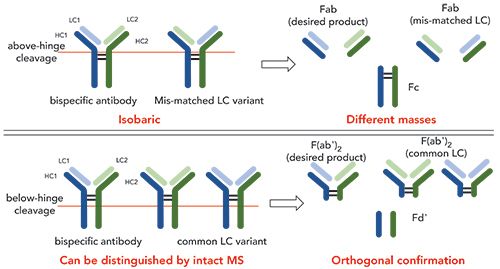
Both common LC species (Figure 1) in bispecific antibody A were detected by MS analysis of the subunits, and the results informed the further development of the purification process to successfully remove both common LC species. This assay is suitable for in-process pool and drug substance samples from bispecific antibody A. Mass spectrometry–based relative quantitation can also be performed by comparing the sample to an assay control that contains a known level of the mismatched LC variant.

Figure 1: Overlay of deconvoluted mass spectra showing the masses of bispecific antibody A heterodimer with two different common LC variants. Other unlabeled masses are due to expected PTMs of the heterodimer.
Method Qualification for an Ion-Exchange Chromatography–Based Assay for Homodimer Quantitation
To facilitate homodimer removal, bispecific antibody B was specifically engineered so that the two homodimers have different pI values. As a result, an ion exchange chromatography (IEC)–based method employing a pH gradient was developed to monitor homodimers.
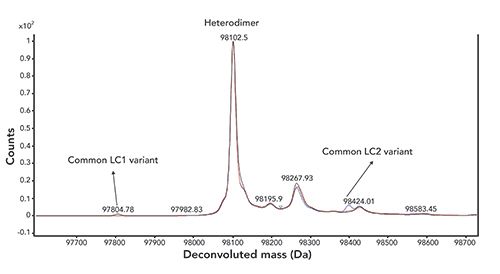
This IEC method is suitable for monitoring in-process pool and drug substance samples (Figure 2). Specificity, sensitivity, linearity, precision, accuracy, and autosampler stability were successfully assessed during method qualification. The specificity of the method was demonstrated with no interference was observed by comparing the sample spiked with purified homodimer 1 with formulation buffer. The limit of detection (LOD) of the method is 2% with a wide linear range up to 10% (w/w, relative to the heterodimer). The precision of the method was evaluated by two analysts, each making triplicate sample preparations and then running on two instruments using two column batches. The relative standard deviation (RSD) value was 2.0% for the measurement of homodimer 1 concentration relative to the heterodimer. The accuracy of the method was assessed by preparing bispecific antibody B at three concentration levels (50%, 100%, and 150% of target concentration) and spiking homodimer 1 at five concentration levels in formulation buffer (0, 1%, 2%, 5%, and 10%) before injecting each in triplicate. The recoveries for bispecific antibody B and spiked homodimer 1 were observed at 94.9–106.2% and 95.9–99.1%, respectively. In addition, 24 h autosampler stability was demonstrated as no difference in the chromatograms were observed for the initial time-point compared to the 24 h time-point samples.

Figure 2: Overlaid chromatograms of bispecific antibody B in (a) protein A process pool, (b) UF/DF pool, (c) analytical standard, and (d) formulation buffer.
During the development of the charge heterogeneity method for heterodimer, we observed strong binding of the heterodimer to the cation-exchange chromatography resin. These data indicated the possibility of a positive charge patch on the surface of bispecific antibody B heterodimer. Given that homodimer 1 contains two mutations with negatively charged amino acid residues, a potential range of linearity for homodimer 1 in the presence of the heterodimer was explored. Because homodimer 2 was fully removed by the purification process (determined by a CE-based assay [data not shown]), a similar assessment was not pursued.
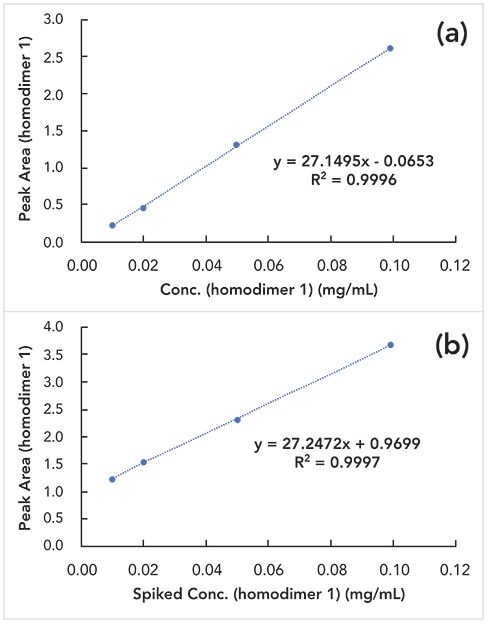
In this experiment, we compared the signal-concentration response curves of the purified homodimer 1 spiked from 0 to 10% (w/w) into two samples: formulation buffer and a bispecific antibody B analytical standard. The peak area versus homodimer 1 concentration plots are shown in Figure 3. The coefficient of determination (R2) was greater than 0.999 and the recovery was between 90 and 110% for all levels of homodimer 1 spiked in both formulation buffer and analytical standard. Specificity was further demonstrated by showing that the heterodimer does not interfere with the measurement of homodimer 1 within the studied range. The presumed positive charge patch on the heterodimer does not appear to result in specific charge–charge interactions between the heterodimer and homodimer 1. Therefore, the IEC method is suitable to quantify the homodimer 1 of the engineered bispecific antibody B.

Figure 3: Linearity of the homodimer 1 in the presence of heterodimer. (a) Homodimer 1 peak area versus concentration plot using purified homodimer 1 spiked in formulation buffer; (b) homodimer 1 peak area versus concentration plot using purified homodimer 1 spiked into the heterodimer analytical standard.
Conclusions
Bispecific antibodies provide new possibilities for drug development along with unique challenges in analytical method development. None of the current manufacturing and purification strategies can ensure 100% heterodimerization, thus analytical methods to detect and measure the levels of undesired product variants are required for the development of bispecific antibodies. Given the unique properties of the product variants, such as chain-pairing variants, product specific analytical methods are often needed. The MS analysis of subunits presented in this article is one of the many possible assay designs. Continued effort from the community is needed to realize further advances in this area. One of the possible directions is to improve the chromatographic resolution of the subunits to allow more accurate quantitation of the variants. Different modes of separation and multidimensional separation can also be explored for this purpose.
On the other hand, the design of bispecific antibodies often involves careful protein engineering. Such engineering requires scientists who are involved in the drug development process to carefully consider the possible impact on the physicochemical properties of the molecule. To ensure the suitability of an assay, assay developers need to anticipate the potential impacts to all aspects of assay performance, and design qualification plans accordingly.
Acknowledgments
The authors want to thank Cinzia Stella for providing the opportunity to share our knowledge in this field, and thank Michelle Beardsley for presenting the interesting case of bispecific antibody A to us. We also want to thank Richard Seipert, Jason Gruenhagen, Lu Dai, and Matt Kalo for providing critical review for this article.
References
- L Yu and J. Wang, Cancer Res. Clin. Oncol.145(4), 941–956 (2019).
- W.R. Strohl, Protein & Cell.9(1), 86–120 (2018).
- S.E. Sedykh, V.V. Prinz, V.N. Buneva, and G.A. Nevinsky, Drug Design Devel. Therapy12, 195–208 (2018).
- Dahlén, N. Veitonmäki, and P. Norlén, Ther. Advances Vaccines Immunotherapy 6(1), 3–17. 2018;
- R.E. Kontermann and U. Brinkmann, Drug Discov. Today.20(7), 838–847 (2015).
- Riethmüller, Cancer Immunity12, 12 (2012).
- Nisonoff and M.M. Rivers, Arch. Biochem. Biophys.93(2), 460–462. 1961;
- U. Brinkmann and R.E. Kontermann, mAbs9(2), 182–212 (2017).
- G.L. Moore, M.J. Bernett, R. Rashid, E.W. Pong, D.-HT. Nguyen, and J. Jacinto, et al., Methods 154, 38–50 (2019).
- G. Giese, A. Williams, M. Rodriguez, and J. Persson, Biotechnol. Progr.34(2), 397–404 (2018).
- A.D. Tustian, C. Endicott, B. Adams, J. Mattila, and H. Bak, mAbs8(4), 828–838 (2016).
- S.M. Lewis, X. Wu, A. Pustilnik, A. Sereno, R. Huang, H.L. Rick, et al., Nat. Biotechnol.32, 191 (2014).
- T.S. Von Kreudenstein, E. Escobar-Carbrera, P.I. Lario, I. D’Angelo, K. Brault, J. Kelly, et al., mAbs 5(5), 646–654 (2013).
- W. Shatz, S. Chung, B. Li, B. Marshall, M. Tejada, W. Phung, et al., mAbs 5(6), 872–881 (2013).
- K. Gunasekaran, M. Pentony, M. Shen, L. Garrett, C. Forte, A. Woodward, et al., J. Biological Chem.285(25), 19637–19646 (2010).
- W. Dall’Acqua, A.L. Simon, M.G. Mulkerrin, and P. Carter, Biochemistry 37(26), 9266–9273 (1998).
- Husain and D. Ellerman, BioDrugs 32(5), 441–464 (2018).
- M. Dillon, Y. Yin, J. Zhou, L. McCarty, D. Ellerman, D. Slaga, et al. mAbs9(2), 213–230 (2017).
- W. Schaefer, J.T. Regula, M. Bähner, J. Schanzer, R. Croasdale, H. Dürr, et al. Proc. Natl. Acad. Sci. USA. 108(27), 11187–11192 (2011).
- C.L. Young, Z.T. Britton, and A.S. Robinson, Biotechnol. J. 7(5), 620–634 (2012).
- L. Schachner, G. Han, M. Dillon, J. Zhou, L. McCarty, D. Ellerman, et al., C. Anal. Chem.88(24), 12122–12127 (2016).
- L.K. Kimerer, T.M. Pabst, A.K. Hunter, and G. Carta, J. Chromatogr. A 1601, 133–44 (2019).
- S. Wang, A.P. Liu, Y. Yan, T.J. Daly, N. Li, J. Pharm. Biomed. Anal. 154, 468–475 (2018).
- Wang, B. Vemulapalli, M. Cao, D. Gadre, J. Wang, A. Hunter, et al., mAbs 10(8), 1226–1235 (2018).
- M. Cao, C. Wang, W.K. Chung, D. Motabar, J. Wang, E. Christian, et al., mAbs 10(8), 1236–1247 (2018).
- R.J. Woods, M.H. Xie, T.S. Von Kreudenstein, G.Y. Ng, and S.B. Dixit, mAbs 5(5), 711–722 (2013).
- F.D. Macchi, F. Yang, C. Li, C. Wang, A.N. Dang, J.C. Marhoul, et al., Anal. Chem. 87(20), 10475–10482 (2015).
Ming Lei is with the Protein Analytical Chemistry group and Tao Chen is with the Small Molecule Analytical Chemistry group, both at Genentech, Inc., in South San Francisco, California. Direct correspondence to: lei.ming@gene.com.













, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

