Ferritin as a Natural Protein Scaffold: Building a Multivalent Ferritin–Fab Conjugate
Special Issues
In this study, a conjugation strategy for Fab–ferritin conjugates, used for drug delivery, was successfully optimized using LC–MS. Characterization of the resulting conjugates was performed using SEC-MALS-QELS.
The design of molecules with a high degree of valency can be useful for receptor clustering, T-cell recruiting, agonist activation, and half-life extension. Although this is traditionally accomplished using polymer-based molecular scaffolds, biocompatibility and the fate of polymer by-products are often of concern. Multivalent, self-assembling scaffolds such as ferritin, a ubiquitous protein found in most human cell types in addition to invertebrates, higher plants, fungi and bacteria, offer an attractive “natural” alternative to polymer-based scaffolds. In mammals, ferritins are composed of 24 subunits that form an icosahedron with an external hydrodynamic radius of 6 nm, and an overall molar mass of approximately 474 kDa, depending on the biological tissue from which it is derived. The utility of molecular cages, such as ferritin and its derivatives for applications in drug delivery, is well-known. Here, we describe the design, production, and characterization of a multimeric ferritin–antibody fragment conjugate. Following optimization of the conjugation strategy using LC–MS, in-depth characterization was performed using SEC-MALS-QELS. The combined results of this study confirmed that Ferritin–Fab conjugates were successfully generated.
There is a growing trend within the field of biotherapeutics to develop molecules with a high degree of valency (1,2). Ferritin family proteins, found ubiquitously in nature, possess many attractive features for the delivery of biotherapeutics (3–6). Ferritin is made up of 24 subunits held together by non-covalent interactions, arranged in an icosahedral cage with 2, 3, and 4-fold axes of symmetry around a central cavity (7,8). While, in nature, the ferritin cavity is used to store and enucleate iron, it can also be repurposed for the encapsulation of drugs or dyes (9). Below pH 3, the ferritin assembly may then be dissociated and reconstituted (10), enabling drugs to be trapped inside the protein “cage” (11). Encapsulation of molecules such as cisplatin (12,13), carboplatin (14), oxaliplatin (15), ruthenium (16,17), and gold (18,19) has been achieved using various ferritin cage disassembly or reassembly procedures. More recently, it was used for the delivery of cisplatin, but without the use of an encapsulation protocol (20). In this case, the interaction was attained through ligand-metal chemistry (21,22), with the preparation of a 1:1 complex between cisplatin and the monomeric subunits of the human ferritin cage (11). Thus, ferritin proteins are attractive vectors for the delivery of drug molecules, either through encapsulation in the central cavity, or through display of drug molecules on the external surface (23). The latter can be achieved using molecular engineering techniques, including the addition of a peptide or protein tag (24).
However, to the authors’ knowledge, there are no examples of adapting ferritin for delivery of multimerized antibody fragment (Fab) therapeutics via chemical conjugation. Here, we describe a drug delivery system using ferritin to multimerize a Fab therapeutic. Commercially available horse spleen ferritin (HSF), with its iron core removed (Millipore Sigma SKU# A3641), was chemically linked through solvent exposed native amino acids to a human Fab. The conjugation strategy was optimized based on analysis, using liquid chromatography in-line with a mass spectrometry detector (LC–MS), while characterization of the resulting conjugates was performed using size exclusion chromatography (SEC) in-line with a multi-angle light scattering detector (MALS) and quasi-elastic light scattering detector (QELS). The results of this study confirmed Fab was successfully conjugated to the HSF cage via site-specific conjugation of the C-terminal cysteine (Cys) on the Fab to solvent exposed lysines on HSF.
Results and Discussion
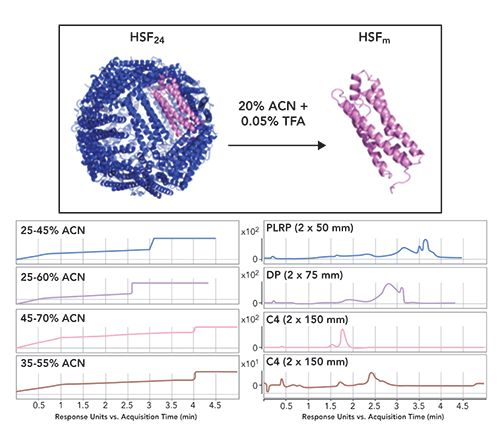
The first step in developing the conjugation protocol was optimization of the reversed-phase method to enable monitoring of the conjugation reactions. Three different reversed-phase columns and four different gradient conditions were scouted (Figure 1). Based on this analysis, a C4 column (Waters, Acquity UHPLC Protein BEH C4) with a 35%–55% acetonitrile (ACN) gradient resulted in optimal baseline resolution of ferritin (around 2.5 min) from the other impurities in the sample, predominantly the contaminant at around 1.75 min (Figure 1, bottom right panel). While chromatographic separation was achieved, the unambiguous identification of the earlier eluting contaminant was not accomplished. However, the deconvoluted mass from the LC–MS spectrum suggested that it was related to HSF (data not shown).


Figure 1: Top panel shows the structure of the assembled icosahedral cage (HSF24) and the denaturing conditions used for its dissociation into the monomer (HSFm). The lower panel compares gradients and corresponding chromatographic traces of the absorbance at 280 nm for the reversed-phase conditions tested. Note: ACN is acetonitrile, TFA is trifluoroacetic acid, PLRP is is a rigid macroporous styrene and divinylbenzene (PS/DVB) HPLC phase, DP represents a diphenyl (DP) bonded phase, and C4 refers to hydrophobic alkyl chain length of the stationary phase.
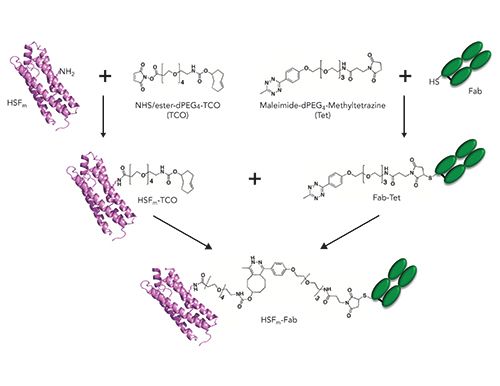
HSF and Fab were conjugated using a two-step procedure: the first involved attachment of a heterobifunctional linker to each component, and this was followed by secondary conjugation using copper-free click chemistry. In each reaction, the reversed-phase method described above was used to characterize intermediate products (Figure 2). All reactions were incubated at room temperature for 18–24 h. To activate HSF, we chose a heterobifunctional N-hydroxysuccinimide (NHS), trans-cyclooctene (TCO), PEG4 linker (NHS-TCO, Figure 2)(BroadPharm, BP-22418). Three different molar excesses of linker (3:1, 20:1, 100:1) relative to HSF monomer (HSFm) were evaluated and monitored by LC–MS (Figure 3a–3d) to generate the HSF displaying a TCO functional group (HSF-TCO).

Figure 2: Schematic representation of reactions to form the HSFm–Fab conjugate.
Surprisingly, the addition of increasing amounts of the NHS-TCO to HSF resulted in decreased area (280 nm) and broadening of the HSF peak (Figure 3b–3d). Although it is not shown here, the decrease in signal was accompanied by suppression of ionization. One hypothesis to explain these findings pertains to the modified lysines. It is possible that the lysines implicated in the conjugation reaction make solvent contacts that are important for ionization, and the decrease in signal is related to increased conjugation of these lysines. Despite this issue, deconvoluted masses were still obtained, which allowed for characterization of the conjugation reaction.

Figure 3: Reversed-phase-HPLC–MS of HSF-Fab conjugation reactions. Left panels show traces for absorbance (280 nm) where peaks being used for deconvolution are boxed in black, while right panels show corresponding deconvoluted masses (zoomed around masses of interest) for (a) HSFm alone, reaction of HSFm with NHS-TCO at a ratio of (b) 3-, (c) 20-, or (d) 100-fold excess TCO linker, (e) reaction of Fab with 3-fold excess of Mal-Tet linker and (f) SPAAC reaction of HSFm-TCO with Tet-Fab.
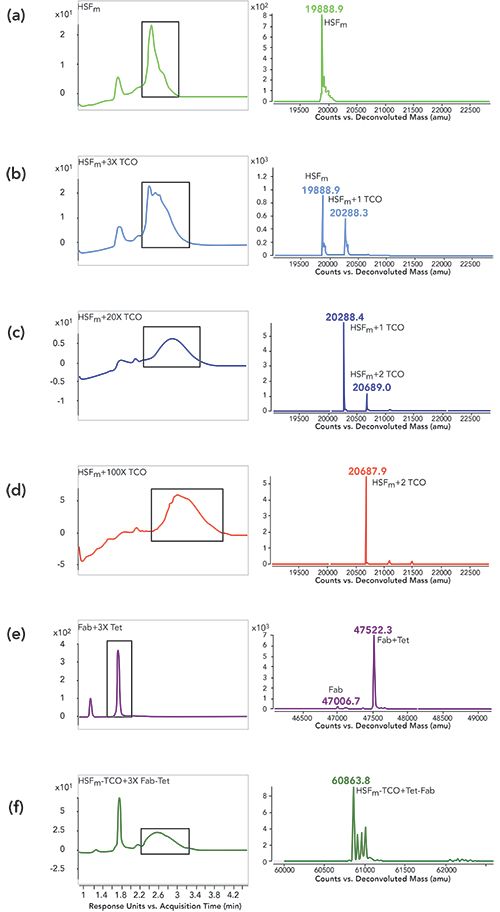
As shown in the deconvoluted mass analysis in Figure 3b, a mixture of unreacted HSFm and one NHS-TCO addition was detected at a reaction ratio of 3:1, while 1-2 additions were detected at a ratio of 20:1 (Figure 3c), and 2 additions were detected at a ratio of 100:1 (Figure 3d). Previously, Zeng and associates identified the presence of surface exposed labile lysines on HSF (25). They found that when a small NHS-alkyne molecule was conjugated to HSF, up to four small linkers were successfully added. However, when lysine reactivity was investigated using a bulky fluorescent probe (5-carboxyfluorescein N-hydroxysuccinimidyl ester) or when saturation of the multiple alkyne moieties was attempted using 3-azido-7-hydroxycoumarin, only one unit per monomer could be detected. In their study, the incomplete modification of HSFm was attributed to steric hinderance since a 200-fold excess of each linker was being added in each reaction, which is consistent with the results reported here. Based on these combined findings, the 20:1 ratio was chosen because it yielded the highest conjugation efficiency of one linker per HSFm.
In parallel with HSF-TCO synthesis, Fab containing a C-terminal Cys was conjugated under physiological conditions to a heterobifunctional maleimide (Mal), methyltetrazine (Tet), PEG4 linker (Mal-Tet, Figure 2) (BroadPharm, BP-22436). The reactivity and lability of the Fab C-terminal Cys was previously reported (26); a 3-fold molar excess of the similar maleimide linkers is sufficient for complete reaction of the Fab Cys (2,27), which was confirmed for the Mal-Tet conjugation to generate the Tet-containing Fab (Fab-Tet) (Figure 3d). In the left panel, two peaks are present, where the peak with retention time around 1.3 min is Fab light chain that has dissociated under denaturing conditions because of a lack of disulfide pairing with the heavy chain, while the peak with retention time around 1.8 min corresponds to the properly assembled Fab. In the right panel, the predominant deconvoluted mass confirms close to complete modification of the Fab with Mal-Tet linker. Subsequent to activation of each molecule, unreacted linker was removed using SEC purification. (GE Healthcare, Superdex 200).
HSF-TCO was then conjugated to Tet-Fab (1:3) using strain-promoted azide-alkyne cycloaddition (SPAAC, Figure 2, 3e) (28–30). In the left panel of Figure 3e, two peaks are observed in the conjugation reaction, where the first peak corresponds to the unconjugated Fab-Tet (1.8 min retention time) and the second corresponds to the HSF-Fab conjugate (2.6 min retention time). In the right panel, the conjugate peak deconvoluted to a mass in-line with expectation. Additional peaks were also observed with an increase of 16–18 Da relative to calculated mass, which are likely water additions on HSF.
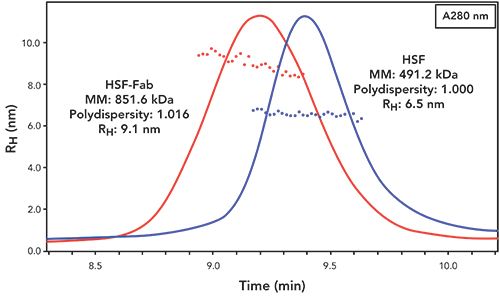
In addition to LC–MS analysis, the assembled HSF-Fab conjugate was characterized under native conditions by SEC-MALS-QELS (Figure 4). As shown in Figure 4, the SEC-MALS-QELS data support a well-behaved soluble conjugate, as confirmed by the symmetrical peak shape and lack of detectable aggregation, as well as an increase in molar mass (MM) and hydrodynamic radius (RH) relative to the unconjugated HSF cage. Although the desired product was confirmed, the difference in MM between the HSF-TCO-Tet-Fab (851.6 kDa) and unconjugated HSF (491.2 kDa) suggests that not all 24 HSFm-TCO subunits were saturated with Fab-Tet. The difference in MM between HSF-Fab and unconjugated HSF suggests an average of 7.7 Fabs per HSF cage, which is consistent with the steric hinderance issues observed by Zeng and associates (25). In that study, a 200-fold excess was used whereas in this work, only a 3-fold excess of Fab-Tet was added to the reaction. Perhaps with a greater excess of Fab-Tet, conjugation to all 24 subunits might be achieved. However, it is likely that there is too much steric crowding to allow for conjugation of 24 Fabs using this approach.

Figure 4: Hydrodynamic radius versus time: native analysis using SEC-MALS-QELS. Chromatogram of absorbance at 280 nm with RH calculated from QELS overlaid across each peak. In addition, averaged molar mass (MM), polydispersity and RH values for HSF and HSF-Fab conjugate are reported.
Materials and Methods
LC–MS analysis
Data was acquired as described previously (31), using an Agilent 1290 Infinity UPLC in tandem with an Agilent 6230 electrospray ionization time-of-flight (UPLC-ESI-TOF) mass spectrometer, operating in positive ion mode. For all gradients, mobile phase A consisted of 0.05% trifluoroacetic acid (TFA) in water while mobile phase B consisted of 0.05% TFA and 100% acetonitrile (Figure 1).
SEC-MALS/QELS Analysis of HSF and HSF-Fab Conjugate
Purity, molar mass (MM), and hydrodynamic radius (RH) were determined using an Acclaim SEC-1000 analytical SEC column run on a Dionex UltiMate 3000 UPLC (Thermo Fisher Scientific), with isocratic elution in phosphate buffered saline (PBS) spiked with an additional 150 mM sodium chloride. In line with ultraviolet (UV) detection, a multi-angle laser light scattering (MALS) detector (Wyatt Instruments), was used to determine molar mass due to Brownian motion, as well as an attached quasi-elastic light scattering (QELS) detector (Wyatt Instruments), to capture fluctuations in intensity of laser light scattered, allowing diffusion coefficients to be measured. Assuming a spherical shape, the Stokes-Einstein relationship was used to calculate RH from D.
Conclusions
This work demonstrates the design and production of a multivalent HSF-Fab conjugate, which presents many benefits as a nanotechnology platform. As a discrete entity, it is a simpler system relative to other delivery technologies such as large polydisperse polymers or liposomes. Moreover, since it is found ubiquitously in living organisms, it is inherently biocompatible, and is easily engineered (32–34). In a recombinant protein format, it has the potential to provide many quality attributes beneficial for downstream development and it is compatible with traditional protein purification techniques to take advantage of well-established manufacturing processes.
This strategy also provides flexibility in making bioconjugates. By using bifunctional click chemistry linkers, virtually any two biomolecules can be conjugated together. Furthermore, the modular approach involving derivatization of each component separately, allows for control of purity at each step prior to clicking the partners together to form the final conjugate. In future work, we hope to explore longer PEG spacers with the goal of increasing conjugation efficiency, and engineering the HSF protein to install site-specific conjugation handles and avoid heterogeneous lysine conjugation.
References
- S. Bhaskar and S. Lim, NPG Asia Mater 9, e371–e389 (2017). doi: 10.1038/am.2016.128.
- W. Shatz, P.E. Hass, N. Peer, M.T. Paluch, C. Blanchette, G. Han, W. Sandoval, A. Morando, K.M. Loyet, V. Bantseev, H. Booler, S. Crowell, A. Kamrath, J.M. Scheer, and R.F. Kelly, PLoS ONE 14:e0218613–20 (2019). doi: 10.1371/journal.pone.0218613.
- J.Y. Li, N. Paragas, R.M. Ned, A. Qiu, M. Viltard, T. Leete, I.R. Drexler, X. Chen, S. Sanna-Cherchi, F. Mohammed, D. Williams, C.S. Lin, K.M. Schmidt-Ott, N.C. Andrews, and J. Barasch, Dev. Cell16(1), 35–46 (2009). doi: 10.1016/j.devcel.2008.12.002.
- L. Li, C.J. Fang, J.C. Ryan, E.C. Niemi, J.A. Lebrón, P.J. Björkman, H. Arase, F.M. Torti, S.V. Torti, M.C. Nakamura, and W.E. Seaman, PNAS107(8), 3505–3510 (2010). doi: 10.1073/pnas.0913192107.
- S. Zhang, J. Zang, H. Chen, M. Li, C. Xu, and G. Zhao, Small13, 1701045–6 (2017). doi: 10.1002/smll.201701045.
- D. He and J. Marles-Wright, N. Biotechnol.32, 651–657 (2015). doi: 10.1016/j.nbt.2014.12.006.
- P.M. Harrison, Biochem. Ed.14, 154–162 (1986). doi: 10.1016/0307-4412(86)90203-7.
- P.M. Harrison and P. Arosio, Biochim. Biophys. Acta1275, 161–203 (1996). doi: 10.1016/0005-2728(96)00022-9.
- S. Ciambellotti, A. Pratesi, M. Severi, G. Ferraro, E. Alessio, A. Merlino, and L. Messori, Dalton Trans. 47, 11429–11437 (2018). doi: 10.1039/C8DT00860D.
- J.M. Domínguez-Vera and F. Colacio, Inorg. Chem. 42, 6983–6985 (2003). doi: 10.1021/ic034783b.
- X.T. Ji, L Huang, and H-Q. Huang, J Proteomics75, 3145–3157 (2012). doi: 10.1016/j.jprot.2012.03.013.
- Z. Yang, X. Wang, H. Diao, J. Zhang, H. Li, H. Sun, and Z. Guo, Chem. Comm. 3453–3455 (2007). doi: 10.1039/B705326F.
- N. Pontillo, F. Pane, L. Messori, A. Amoresano, and A. Merlino, Chem. Comm.52, 4136–4139 (2016). doi: 10.1039/C5CC10365G.
- N. Pontillo, G. Ferraro, J.R. Helliwell, A. Amoresano, and A. Merlino, ACS Med. Chem. Lett.8, 433–437 (2017). doi: 10.1021/acsmedchemlett.7b00025.
- R.Xing, X. Wang, C. Zhang, Y. Zhang, Q. Wang, Z. Yang, and Z. Guo, J. Inorg. Biochem. 103, 1039–1044 (2009). doi: 10.1016/j.jinorgbio.2009.05.001.
- K. Fujita, Y. Tanaka, T. sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, and S. Kizaka-Kondoh, J. Am. Chem. Soc. 136, 16902–16908 (2014). doi: 10.1021/ja508938f.
- Y. Takezawa, P. Böckmann, N. sugi, Z. Wang, S. Abe, T. Murakami, T. Hikage, G. Erker, Y. Wantanabe, S. Kitagawa, and T. Ueno, Dalton Trans.40, 2190–2195 (2011). doi: 10.1039/c0dt00955e.
- D.M. Monti, G. Ferraro, G. Petruk, L. Maiore, F. Pane, A. Amoresano, M.A. Cinellu, and A. Merlino, Dalton Trans.46, 15354–15362. doi: 10.1039/C7DT02370G (2017).
- G. Ferraro, D.M. Monti, A. Amoresano, N. Pontillo, G. Petruk, F. Pane, M.A. Cinellu, and A. Merlino, Chem. Comm.52, 9518–9521. doi: 10.1039/C6CC02516A (2016).
- G. Ferraro, S. Ciambellotti, L. Messori, and A. Merlino, Inorg. Chem. 56, 9064–9070. doi: 10.1021/acs.inorgchem.7b01072 (2017).
- L. Messori and A. Merlino, Coord. Chem. Rev.315, 67–89. doi: 10.1016/j.ccr.2016.01.010 (2016).
- T.G. Appleton, Coord. Chem. Rev. 166, 313–359. doi: 10.1016/S0010-8545(97)00047-7 (1997).
- M. Kanekiyo, C-J. wei, H.M. Yassine, P.M. McTamney, J.C. Boyington, J.R.R. Whittle, S.S. Rao, W-P. Kong, L. Wang, and G.J. Nabel, Nature 499, 102–106 (2013). doi: 10.1038/nature12202.
- J.M. Hooker, A. Datta, M. Botta, K.N. Raymond, and M.B. Francis, Nano. Lett. 7, 2207–2210 (2007). doi: 10.1021/nl070512c.
- Q. Zeng, R. Reuther, J. Oxsher, and Q. Wang, Bioorg. Chem. 36, 255–260 (2008). doi: 10.1016/j.bioorg.2008.06.001.
- S. Jevševar, M. Kusterle, and M. Kenig, in Site-Specific Protein Labeling, P. Chames, Ed. (Humana Press, Totowa, New Jersey, 2012), pp 233–246.
- W. Shatz, P.E. Hass, M. Mathieu, H.S. Kim, K. leach, M. Zhou, Y. Crawford, A. Shen, K. wang, D.P. Chang, M. Maia, S.R. Crowell, L. Dickmann, J.M. Scheer, and R.F. Kelley, Mol. Pharm. 13(9), 2996–3003 (2016). doi: 10.1021/acs.molpharmaceut.6b00345.
- N.J. Agard, J.A. Prescher, and C.R. Bertozzi, J. Am. Chem. Soc.126, 15046–15047 (2004). doi: 10.1021/ja044996f.
- J.M. Baskin, J.A. Prescher, S.T. laughlin, N.J. Agard, P.V> Chang, I.A. Miller, A. Lo, J.A. Codelli, and C.R. Bertozzi, PNAS104, 16793–16797 (2007). doi: 10.1073/pnas.0707090104.
- J. Dommerholt , F.P.J.T. Rutjes, and F.L. van Delft, Top Curr. Chem. (Cham) 374, 16–20 (2016). doi: 10.1007/s41061-016-0016-4.
- J. Zarzar, W. Shatz, N. Peer, R. Taing, B. McGarry, Y. Liu, D.G. Greene, and I.E. Zarraga, Biophys. Chem.236, 22–30 (2017). doi: 10.1016/j.bpc.2017.10.003.
- T. Treffry, P.M. Harrison, A. Luzzago, and G. Cesareni, FEBS Letters247, 268–272 (1989).
- P. Santambrogio, S. Levi, A. Cozzi, E. Rovida, A. Albertini, and P. Arosio, J. Biol. Chem.268, 12744–12748 (1993).
- P. Rucker, F.M. Torti, and S.V. Torti, Protein Eng., Des. Sel. 10(8) 967–973 (1997). doi: org/10.1093/protein/10.8.967.
Whitney Shatz, Remo Perozzo and Yogeshvar N. Kalia are with the School of Pharmaceutical Sciences at the University of Geneva, in Geneva, Switzerland. Shatz, Craig Blanchette, Patrick Holder, and Robert F. Kelley are with Genentech, in South San Francisco, California. Direct correspondence to: shatz.whitney@gene.com















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

