Troubleshooting LC Separations of Biomolecules, Part II: Passivation and Mobile-Phase Additives
LCGC North America


If you are analyzing metal-sensitive biomolecules, and a bioinert instrument is unavailable, or insufficient, passivation or mobile-phase additives may help. Here’s how to use those solutions, with tips for avoiding potential pitfalls.
Bioinert and biocompatible liquid chromatography (LC) systems are becoming more commonplace in laboratories, but the majority of biomolecule separations still use LC systems composed primarily of stainless steel parts. Can passivation or mobile phase additives improve separations on these systems for metal-sensitive biomolecules?
In the March, 2020 installment of “LC Troubleshooting,” we started a series of articles on the topic of biomolecule separations, and discussed the use of biocompatible components, including column hardware and parts of the liquid chromatography (LC) flow path (1). Bioinert high performance liquid chromatography (HPLC) systems and column hardware are great potential solutions for the analysis of biomolecules that are sensitive to metal interaction. However, as a user, you may not have access to these systems, and, even if you do, it is prudent to anticipate separation problems that might arise due to different materials in the system. For example, a recent study of an iron-free HPLC system that utilized titanium pump heads documented the negative effect of leached titanium ions on peak shape and retention of metal-sensitive analytes (2). Therefore, it is best to utilize a system suitability test (for example, a mixture of standards that contains metal-sensitive compounds) that not only reflects the health of the chromatography system, but is also relevant to the molecules you are analyzing. Importantly, even among nominally identical HPLC systems, the overall level of metal interaction that a sensitive analyte might experience is variable, depending on the condition of the system components themselves. In this article, we discuss mitigation strategies for traditional stainless steel HPLC systems when bioinert versions are either unavailable or insufficient for the analysis of
metal-sensitive biomolecules.
What Can Be Done About Issues Related to Bioinertness?
Stainless steel (SS) and other chromium-based alloys are widely used materials in many industries, including the construction, automotive, energy, food, aerospace, and biomedical fields. Although SS is a rust-resistant alloy, passivation remains a critical step in maximizing the corrosion resistance of components and parts fabricated from SS (3–5). Passivation is typically the first procedure implemented for improving the chromatography of metal-sensitive biomolecules using SS-containing HPLC components. In general, the term passivation describes any treatment of a metal surface that removes prior corrosion and prevents future corrosion for some period of time. There are different grades of SS, and the quality of the SS directly affects its resistance to corrosion. The two most utilized grades of SS are 304 and 316, with various types of 316 SS material utilized for different purposes. Grade 304 contains 18% chromium and 8% nickel, while grade 316 contains 16% chromium, 10% nickel, and 2% molybdenum. Importantly, the inclusion of the molybdenum alloy in grade 316 SS improves its corrosion resistance compared to grade 304 SS, particularly against saline- or chloride-containing solutions. However, even for high quality SS, corrosion can still occur at levels sufficient to cause trouble for bio-separations (6–9). Passivation procedures for SS usually involve chemical treatment that removes surface rust while maintaining alloyed chromium. Nitric, phosphoric, or citric acid are the typical chemicals of choice for passivation procedures. During the passivation procedure, a protective chromium oxide (Cr2O3) layer is formed on the SS surface, and the iron at the surface is dissolved selectively (10). The concentration of Cr2O3 and the depth of the protective layer depend on the acid concentration, temperature, and duration of the passivation step (11). In a study that compared nitric acid, citric acid, and Citrisurf (a commercially available citrate-based passivation agent) the authors found that the content of iron oxide (Fe2O3) rapidly decreases in the protective layer within the first 2 h of passivation, while chromium oxide content increases (12). The American Society for Testing and Materials (ASTM) International A967 standard, Standardization Specification for Chemical Passivation Treatments for Stainless Steel Parts, is an important industry document that specifies various passivation procedures for SS. Readers interested in learning more about SS passivation procedures will find this standard document to be a rich resource (13).
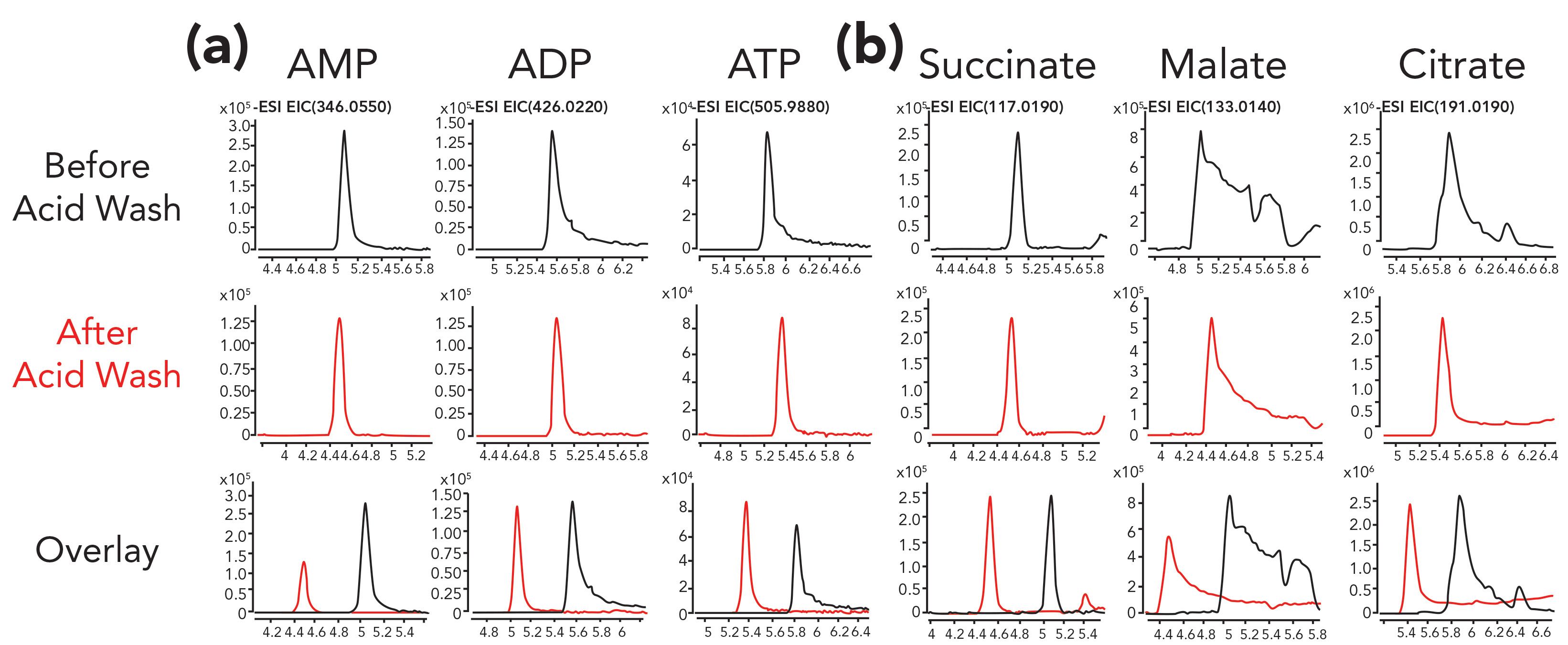
In an initial pilot study of our own, we passivated an HPLC system using 0.5 wt% phosphoric acid in acetonitrile:water (90:10), comparing chromatographic results before and after the acid wash. Here, peak shapes for the small molecule adenosine monophosphate (AMP) were symmetrical prior to passivation. However, for metabolites with more phosphate groups such as adenosine diphosphate (ADP) and adenosine triphosphate (ATP), severely tailed peaks were observed prior to the acid wash. These peaks are all shown in the first row of Figure 1. After overnight flushing using the passivation solution, much sharper peaks were observed for both ADP and ATP. The mass spectrometry (MS) signal for ATP was also slightly higher following the phosphoric acid wash. In contrast, the signal for AMP was reduced after the wash, which could be due to residual phosphoric acid still in the system suppressing the ionization of AMP. Furthermore, the peak shapes for some organic acids (malate and citrate) also improved following the phosphoric acid wash (Figure 1b). However, the peak shapes for malate and citrate, even after the passivation procedure, still showed a lot of tailing, leaving room for further improvement. In this case, additional steps were required to mitigate interactions between the analytes and metals in the HPLC system (14–16).


Figure 1: Separation of (a) phosphorylated nucleotides and (b) organic acids improved after treatment of a conventional SS HPLC system by flushing overnight with a phosphoric acid solution (0.5% phosphoric acid in 90:10 acetonitrile:water) for 16 h at room temperature. Following the phosphoric acid treatment, the HPLC system was flushed with ultrapure water for 1 h to clean the system and avoid introducing phosphoric acid into the mass spectrometer. Gradient HILIC separation was conducted with an aqueous solution of 10 mM ammonium acetate adjusted to pH 9 with ammonium hydroxide in bottle A and acetonitrile:water (90:10) of 10 mM ammonium acetate, pH 9 in bottle B. Figure axes labels are Acquisition Time (min) for x-axis and Counts for y-axis.
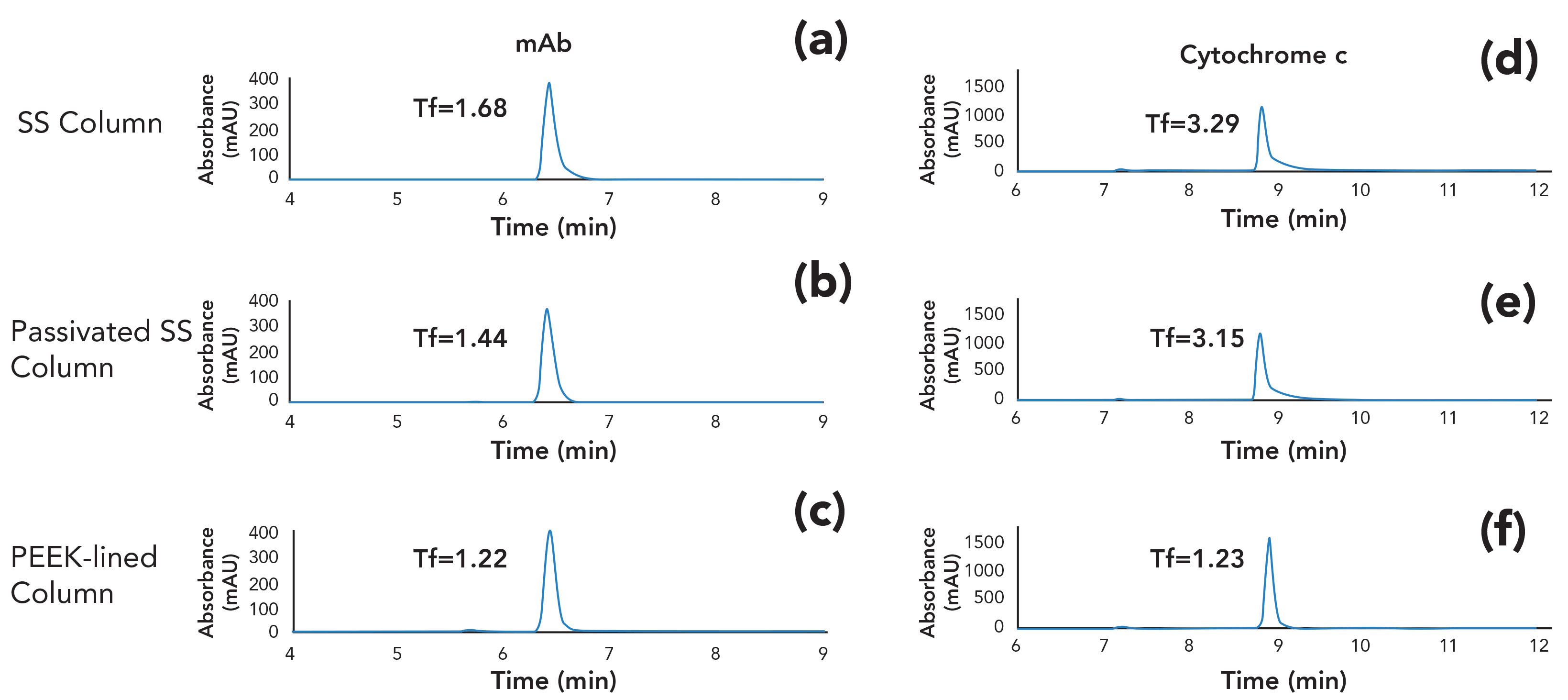
In a different study, we set out to examine whether SS columns could be passivated using the same phosphoric acid washing procedure. It is important to note that not all columns can be safely treated with these conditions, because the stationary phase may not be chemically stable at low pH (17). Please be sure to check the recommendations of the manufacturer regarding the appropriate mobile phase pH range for a given column. In this example, we studied the peak shapes of an intact monoclonal antibody (mAb) and cytochrome c analyzed using a size-exclusion column coupled to a UV detector. After the passivation solution was flowed through the bioinert HPLC system and SS analytical column overnight, a sharper chromatographic mAb peak with reduced tailing factor (Tf) was observed (Figure 2a and 2b). Interestingly, the use of PEEK-lined column hardware loaded with the same SEC particles still yielded the best chromatographic results with the lowest Tf (Figure 2c). This trend becomes more apparent with cytochrome c, (Figure 2d, 2e, and 2f). These results suggest that passivation alone may be insufficient and equipping the experimental setup with bioinert materials (for example, connecting capillaries and column hardware) would yield better results for biomolecules prone to interact with metal surfaces.

Figure 2: Separation of NISTmAb and cytochrome c on SEC columns in (a,d) SS, (b,e) passivated SS, and (c,f) PEEK-lined column hardware. The mobile phase was 150 mM sodium phosphate at pH 7.0.
As described in the previous section, the principle aim of passivation procedures is to remove prior corrosion and prevent future corrosion for some period of time. However, passivation alone may not be enough to facilitate high quality biomolecule separations in situations where parts of an HPLC pump leach trace levels of metals (for example, titanium or iron) into the mobile phase (2,18), or when the sample matrix is contaminated with metals. Different types of metal chelators (compounds that sequester metal atoms) have been used as mobile-phase additives to improve separation performance for analytes that are sensitive to the presence of metals in the analytical system. These metal chelators have been used in a variety of ways including spiking them directly into samples, injecting them into the LC system between sample analyses, and using them as mobile phase additives. When they are used as mobile-phase additives, one must consider the target analytes for each specific application and how the mobile-phase additive could potentially interfere with the analysis. For example, citric acid has been used as a mobile-phase additive to improve the analysis of phosphopeptides in reversed-phase separations (7). Ethylenediaminetetraacetic acid (EDTA) is another well known metal chelator that is effective for improving the peak shape of metal-sensitive metabolites separated using normal-phase chromatography (15,16) and for the separation of monoclonal antibodies using cation-exchange chromatography (CEX) (19). More recently, medronic acid was used as a mobile-phase additive to improve the peak shape and signal for polar metabolites and phosphopeptides (20). In Figure 3, medronic acid is also shown to improve the peak shape of fluorescently labeled N-glycans separated on an amide HILIC column.The later eluting structures each contain two sialic acid groups, giving them a strong tendency to interact with trace amounts of metal in the system.Rather than taking the system offline for passivation, the addition of a very low concentration of medronic acid to the aqueous mobile phase resolves the problem immediately. Furthermore, this benefit persists for at least one week after returning to the use of mobile phase without medronic acid.
Potential Pitfalls of Using Mobile-Phase Additives to Improve Biomolecule Peak Shape
Although mobile-phase additives are an attractive option to yield better bioanalytical results, a few caveats associated with this approach need to be considered.
Ion Suppression
It has been well documented that the use of EDTA as a mobile phase additive is associated with ion suppression effects that reduce the signal of target analytes when hyphenated with MS detection (17,20). When using mobile-phase additives, the benefit is that the passivation of an HPLC system can be continuously maintained, but it is important to keep in mind that the right dose of additive concentration is required for optimal analysis, and this must be determined empirically on a case-by-case basis. For example, use of medronic acid at high concentrations (such as, for instance, 10 µM) can still cause ion suppression, and use of medronic acid at low concentrations (for example, 0.5 and 1 µM) may not yield the optimal chromatographic results (20).
Persistence in the HPLC System
Another important consideration is whether the additive poses a risk to future work by slowly leaching from the LC system, even after switching to mobile phases that do not contain the additive. For example, EDTA has been reported to be retained on reversed-phase columns when used as a mobile-phase additive for phosphopeptide analysis (7). Moreover, due to EDTA’s low solubility under acidic pH conditions (0.1% formic acid), precipitation of EDTA has been observed at the Ionization (ESI) nebulizer needle, and can result in blockage of analytical columns; obviously these are both undesirable outcomes for any LC method. In contrast, citric acid was found to be more amenable to prolonged use as a mobile- phase additive (7).
Summary
In this installment of “LC Troubleshooting,” we have discussed approaches that can improve the analysis of biomolecules prone to strong interaction with metal components in LC systems or trace metals present in the sample flow path. The two methods that we described here include chemical passivation and use of metal chelators as mobile-phase additives. If corrosion or rusting of an LC system is suspected, the system should be passivated. Next, if applicable, mobile-phase additives could be used to chelate any trace metals leached from pump heads or other metal components, or metals present in the sample matrix. Lastly, bioinert instrument components and column hardware still represent the best option for preventing adsorption of proteins on instrument components and column hardware. In particular, the use of bioinert materials where the biomolecules may directly interact with these surfaces is recommended. For example, use of PEEK-lined capillaries between the injector and detector is recommended, as is the use of PEEK-lined column hardware to limit potential metal-interaction sites. Understanding the pros and cons of passivation approaches and mobile-phase additives should support the development of effective and robust methods for accurate and reproducible analysis of biomolecules.
References
- J.J. Hsiao, G.O. Staples, and D.R. Stoll, LCGC N Am. 38(3), 146–150 (2020).
- M. De Pra, G. Greco, M.P. Krajewski, M.M. Martin, E. George, N. Bartsch, and F. Steiner, J. Chromatogr. A 1611, (2020). doi: 10.1016/j.chroma. 2019.460619.
- K.E. Collins, C.H. Collins, and C.A. Bertran, LCGC N Am. 18(6), 600–608 (2000).
- K.E. Collins, C.H. Collins, and C.A. Bertran, LCGC N Am. 18(7), 688–692 (2000).
- R. Shoup and M. Bogdan, LCGC 7(9), 742–744 (1989).
- A. Fleitz, E. Nieves, C. Madrid-Aliste, S.J. Fentress, L.D. Sibley, L.M. Weiss, R.H. Angeletti, and F-Y. Che, Anal. Chem. 85(18), 8566−8576 (2013). doi: 10.1021/ac401691g.
- D. Winter, J. Seidler, Y. Ziv, Y. Shiloh, and W.D. Lehmann, J. Proteome Res. 8(1), 418−424 (2009). doi: 10.1021/pr800304n.
- K. Sandra, J. Vandenbussche, and P. Sandra, LCGC Europe 31(10), 566–571 (2018).
- J.A. Anspach, S. Rao, and B. Rivera, LCGC North Am. 36(6s), 24–29 (2018).
- I. Olefjord, Mater. Sci. Eng. 42, 161–171 (1980). doi: 10.1016/0025-5416(80)90025-7
- D. Wallinder, J. Pan, C. Leygraf, and A. Delblanc-Bauer, Corros. Sci. 41(2), 275–289 (1998). doi: 10.1016/S0010-938X(98)00122-X.
- C. O’Laoire, B. Timmins, L. Kremer, J.D. Holmes, and M.A. Morris, Anal. Lett. 39(11), 2255–2271 (2006). doi: 10.1080/00032710600755363.
- ASTM International ASTM A967/ A967M–13 (2013) Standard Specification for Chemical Passivation Treatments for Stainless Steel Parts, ASTM International, http://www.astm.org/Standards/A967.htm (Accessed 16 April 2020).
- R.E. Birdsall, J. Kellett, Y.Q. Yu, and W. Chen, J. Chromatogr. B 1126–1127, 121773 (2019). doi: 10.1016/j.jchromb.2019.121773.
- K.T. Myint, T. Uehara, K. Aoshima, and Y. Oda, Anal. Chem. 81(18), 7766–7772 (2009). doi: 10.1021/ac901269h.
- J.J. Pesek, M.T. Matyska, and S.M. Fischer, J. Sep. Sci. 34(24), 3509–3516 (2011). doi: 10.1002/jssc.201100607.
- J. Dolan, LCGC North Am. 33, 694–700 (2015).
- M.R. Euerby, C.M. Johnson, I.D. Rushin, and S. Tennekoon, J. Chromatogr. A 705, 229−245, (1995).
- L. Zhang, T.J. Robinson, and B.D. Schmidt, J. Chromatogr. A 1367, 109–117 (2014). doi: 10.1016/j.chroma.2014.09.051.
- J.J. Hsiao, O.G. Potter, T.W. Chu, and H. Yin, Anal. Chem.90(15), 9457–9464 (2018). doi: 10.1021/acs.analchem.8b02100.


Dwight R. Stoll is the editor of “LC Troubleshooting.” Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 60 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com


Gregory Staples leads an R&D team focused on creating and developing separation and sample preparation solutions for biomolecules at Agilent Technologies in Santa Clara, California.


Jordy Hsiaois an R&D scientist at Agilent Technologies in Santa Clara, California.


Te-Wei Chu is an R&D scientist at Agilent Technologies in Santa Clara, California.


Oscar G. Potter is an R&D scientist at Agilent Technologies in Santa Clara, California.














, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.


New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

