Superficially Porous and Monolithic Columns for Ultrafast Separations in HPLC
Special Issues
Due to economic crisis all over the world it is a time of cost-friendly analyses. Superficially porous and monolithic columns are the tools to serve this purpose. These columns are new generation and can be used for ultrafast separations. This article describes the state-of-the-art for these stationary phases for high performance liquid chromatography (HPLC). The emphasis has been placed on their preparation, properties, applications, comparison, and future perspectives. It has been observed that superficially porous columns may be the choice of future for ultrafast separations.
In the current economic scenario persons such as industrialists, academicians, scientists, and regulatory authorities are looking toward fast analyses. Proteomic, genomic, and metabolic research are also in need of ultrafast separations. Therefore, scientists have turned to fast analyses by reducing the particle size of the column. A twofold reduction in particle size can lead to a 1.4-fold (square root of 2) increase in column efficiency, but back pressure increases four times, which is a serious issue and a subject of extensive research (1). Columns with particle sizes smaller than 2.0 μm have been used to decrease analysis time (2–4) but, unfortunately, they exert high back pressure (~15,000 psi), and, hence, require new instruments. Moreover, it is difficult to achieve high speed, reliability, precision and sensitivity — even using ultrahigh-pressure liquid chromatography (UHPLC) — due to tight packing of columns with small particles. To overcome these problems, scientists worked in other directions and two new types of columns capable of providing ultrafast HPLC analyses have been introduced: Monolithic and superficially porous (5–7) columns.



Monolithic Columns
During the last decade, monolithic columns made of a single piece of monolithic silica were introduced as the alternative to particle-based columns. The monolithic columns are biporous, having larger macropores (2.0 μm) allowing high flow rates with low back pressure and smaller mesopores (13.0 nm) that provide high surface area for good efficiency (8). The advantageous features of monolithic silica columns are their ability to independently control the macro and mesopore diameters as well as the silica skeleton diameter. It is possible to perform analyses with high linear flow velocity without reduced separation efficiency. Monolithic columns are also available with organic materials (polymers) providing high-speed separation of polypeptides and proteins in reversed-phase and ion-exchange modes but showed relatively low efficiency for small molecules. A search of the literature indicates several reviews (9–22) of monolithic columns.
Preparation of monolithic silica gel: Tetramethoxysilane (TMOS) and polyethylene oxide (PEO) are a silica source and a polymer for preparing monolithic columns, respectively. Some publications have described the syntheses using different quantities of reactants (22–28). The prepared monolithics are of TMOS, PEO, and chromolith and methacrylate types. The macropore and skeleton diameter can be controlled by PEO while a simultaneous increase in TMOS is responsible for skeleton diameter and lower macropore volume or thinner skeletons and higher macropore volume. Tanaka and colleagues (23,24) developed monolithic columns and derivatized to C18 materials. As per the authors, a subsequent surface modification of C18 monoliths silica led to a reduction of mesopore size and volume affecting the column performance due to different mass transfer kinetics. Chromolith columns are prepared either in a mold or in a fused silica capillary. The resulting silica monoliths (10–15 cm) are covered with PTFE tubing or with PEEK resin to fabricate a column. Basically, methacrylate monolithic columns are useful for developing fast, efficient, and highly productive purification processes for large biomolecules. Some reviews (25,26) are available in the literature on methacrylate monolithics for preparation, characterization, and applications. The enhanced mass transfer properties, pressure and flow, specific permeability, morphological, and structural characteristics are the main features of these columns.


Figure 1: SEM images of monolithic silica gel (27,28).
Properties of monolithic silica gel: Scanning electron microscopy (SEM) images of monolithic silica are shown in Figure 1 (27,28), indicating silica skeletons and pores as co-continuous. The specific surface area, pore volume, median pore radius, porosity, and equivalent particle diameter are 7.19 m2/g, 1.35 mL/g, 750 nm, 64%, and 0.75 μm, respectively. Sugrue and colleagues (29) reported ion exchange properties of monolithic and particle type iminodiacetic acid modified columns. Lubda and colleagues (30) studied ccomprehensive pore structure characterization of silica monoliths with controlled mesopore size and macropore size by nitrogen sorption, mercury porosimetry, transmission electron microscopy, and inverse size exclusion chromatography. The hydrodynamic characteristics of methacrylate monoliths depend upon their structure, that is, porosity, pore size, and pore size distribution, which are controlled by the preparation–polymerization of the monolith. Basically, methacrylate monolithic columns are recognized by short column length and an extremely high surface-to-volume ratio. The pressure drop is the linear velocity, which is directly proportional to pressure drop. Bencina and colleagues (31) described DNA separation and reported that pore size of about 1.5 μm is quite good for this.

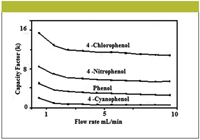
Figure 2: The effect of flow rates on retention factors of phenols on a monolithic column (42).
Applications of monolithic columns: Monolithic columns and capillaries have been used in HPLC and capillary electrochromatography (CEC) due to their remarkable properties. The crucial parameters for high chromatographic performance are not only mesopore and macropore but skeleton diameter as well (32–36). Some computational studies have been carried out to determine the optimum and relative geometries of monolithic columns in HPLC (37,38). Ali and colleagues performed separations of haloperidol (39,40), tadalafil (41), phenols (42), and chloramphenicol (43) in various matrices using a Chromolith RP-18e monolithic column (Merck KGaA, Darmstadt, Germany). The optimization was achieved by adjusting mobile phase compositions, pH, and flow rates. Figure 2 shows effect of the flow rates (1–10 mL/min) on the separation of phenolics in water. The studied phenols were separated successfully at all flow rates (1.0–10.0 mL/min) but the peaks were slightly broad at 1.0- and 2.0-mL/min. flow rates. On the other hand, the detection was poor at flow rates ranging from 4.0 to 10.0 mL/min. Therefore, 3.0 mL/min was selected as the optimum flow rate. Tanaka and colleagues (23,27) compared the pressure exerted by the monolithic column with that of packed columns (Figure 3). The pressure was found to be quite low in monolithics in comparison to others.

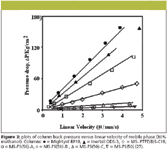
Figure 3
Monolithic capillaries are more effective and efficient for separation due to the absence of the van Deemter A-term contribution. The best results achieved were with a capillary possessing a domain size of 2.2 μm and a corresponding Hmin of 5 μm (44). It is possible to generate 500,000–10,00,000 diffusion limited plate numbers in capillaries of 20–30 cm length in CEC (45). Puy and colleagues (46) synthesized monolithic silica capillaries by a sol-gel process. The effect of skeleton size on the electroosmotic flow (EOF) was investigated with unmodified synthesized monolithic silica capillaries used under various experimental conditions. Ishizuka and colleagues (28) prepared a monolithic capillary and used under electrodriven conditions; indicating better performance in comparison to pressure driven mode. Breadmore and colleagues (47) reported micro-chip capillary electrochromatography in a monolithic silica gel. Gatschelhofer and colleagues (48) used teicoplanin aglycone in monolithic capillary for chiral separation of glycyl-dipeptides. Chankvetadze and colleagues (49) modified monolithic capillary using cellulose tris-(3,5-dimethylphenylcarbamate) and used it for enantioseparations of several drugs. Furthermore, the same group (50) prepared monolithic capillaries covalently bonded with 3,5-disubstituted phenylcarbamate derivatives of cellulose and amylase and used them for chiral separations. The effects of the type of polysaccharides and the substituents, as well as of multiple covalent immobilization of polysaccharide derivatives were studied. While it is not possible to discuss all applications in detail, some selected applications of monolithic phases are given in Table I.

Table I: Applications of monolithic columns and capillaries in HPLC and CEC
Superficially Porous Columns
During the last few years, superficially porous silica particles also sometimes referred to as "shell particles" have evolved that are capable of reducing analysis time up to 70%. The trade name of these columns are Halo, Express, and Poroshell from Advanced Material Technology (Wilmington, Delaware), Supelco (Bellefonte, Pennsylvania), and Agilent Technologies (Palo Alto, California), respectively. During preparation of this article, a thorough search of the literature was carried out and found only a few papers on Express and none on Poroshell 120 columns, while some papers are available on Halo columns. Therefore, Halo columns are discussed further in this article.


Figure 4: SEM images of superficially porous silica particles at different magnifications (6,78).
Figure 4 shows SEM images of superficially porous silica gel particle at different magnifications (6,78) while Figure 5 depicts the cross sections and internal structures of superficially porous particles (6). Normally, superficially porous particles have only a 0.5-μm diffusion path as compared with the 1.5-μm diffusion path of a 3.0-μm totally porous particle. Due to this diffusion path difference, the performance of superficially porous particles becomes more apparent when operating at faster mobile phase flow rates. The silica gel particle has a 2.7-μm overall diameter with porous layer 0.5 μm thick, a 90-Å pore diameter and a 150-m2/g surface area.

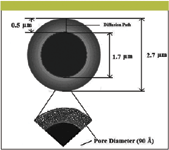
Figure 5: The cross-sectional and internal structures of superficially porous silica gel particle (Halo) (6).
Preparation of shell particles: The first superficially porous silica gel was manufactured in 2000 (78). Small particles with 5.0-μm overall diameter including a 0.25-μm thick porous layer with 30.0-nm pores for the separation of various molecules was developed to take advantage of smaller diffusion distances for low diffusivity solutes. C18 silica gel yielded reduced plate heights, similar to values for totally porous silica gel material. Later, the same author (79) prepared C8 and C18 particles with overall diameter of 2.7 μm with porous layer 0.5 μm thick containing 90 Å as pore diameter. The layer on these particles represents 75% of the volume of the whole, potentially limiting the earlier problems of sample capacity. The silica gel particles of Halo columns consist of a porous shell fused to a solid silica core.
Properties of shell particles: One of the advantages of shell particles includes quick elution of unretained solutes (t0) comparatively to totally porous particles of the same size; resulting in an increase of separation speed by reducing the dead time (about one-half that for comparable columns of totally porous particles). Cunliffe and Maloney (80) carried out a remarkable comparison of shell particles with totally porous silica gel particles (2.0-μm silica particles). As in the case of monolithic columns, the back pressure also was measured at varying flow rates as shown in Figure 6, indicating the lower back pressures of shell particle columns compared with sub-2.0-μm particle size columns. Furthermore, the authors reported 80,000–95,000 theoretical plates just by coupling three 15.0 cm columns together. The back pressure of the coupled columns did not exceed 400 bar at ≤ 1.25 mL/min, enabling the use of traditional HPLC instruments.

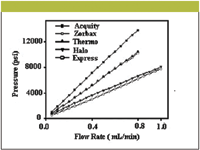
Figure 6: A comparison of pressure and flow rates for five 100 mm à 2.1 mm columns (80). Mobile phase: 50:50 (v/v) waterâacetonitrile.
Guiochon and colleagues carried out a good comparison of shell particle column performance with that of conventional columns (81). As per these authors, the vanDeemter C term measured with shell particle columns was similar or even larger than for fully porous silica particles. The outer structure of the shell particles generates an enhanced film mass transfer resistance, canceling out the benefit of shortening the diffusion path in the particle. Furthermore, the same group (82) compared five different C18 bonded silica gel columns, that is, 3.0- and 5.0- μm Luna, 3.0-μm Atlantis, 3.5-μm Zorbax, and 2.7-μm Halo. The authors separated naphthalene, insulin, and bovine serum albumin as model compounds on these columns at 0.010–3.0 mL/min. flow rates. The C terms of these columns were calculated indicating 2.5 times higher value for the shell particle columns.
The comparisons of plate number, pressure, and plates/bar for shell particle and other columns of smaller particle sizes are shown in Figure 7 (83), which indicates an advantage for shell particle columns in terms of available plates as a function of pressure requirements. As discussed previously, the mass transfer is better in shell particle columns where solutes diffuse more quickly in and out of the porous structure for interaction with the stationary phase. Therefore, for smaller molecules (for example, MW <600) the effect is not dominant. The sample loading capacities of shell particles are quite good in comparison to total porous particles. Almost the same sample loading was observed with 150 and 440 m2/g surface areas of the shell particle column and columns packed with totally porous particles.

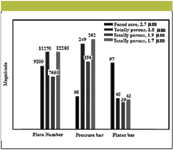
Figure 7: Comparison of plate number, pressure, and plates/bar for sub-2μm fused-core particles (83).
Applications of shell particle columns: Shell particle columns are more useful for delivering over 50% more separating power (theoretical plates) than conventional columns of the same length packed with 3.5-μm totally porous particles (6). Normally, shell particle columns have twice the numbers of theoretical plates and resolving power with low back pressure. The theoretical plates may be achieved up to > 2,00,000/m. The available surface area (150 m2/g) is suitable for separating small molecules with good sample loading capacities while high efficiency is due to narrow particles size distribution and high particle density. The short diffusion paths of the thin porous crust and reduced back pressure allowed mobile phase velocities at the plate height minimum for highest efficiency.
The shell columns are available in C8, C18, reversed-phase amide, and hydrophilic interaction chromatography (HILIC) types and have their own specific properties and applications. Normally, the mobile phases used with these columns are different combinations of acetonitrile and water or acetonitrile and buffers. A wide range of ultrafast shell particle C18 column applications have been reported. Destefano and colleagues (83) used shell particle C18 columns for naphthalene, virginiamycin, pesticides, and explosives separations and reported their baseline resolution within 0.7–3.5 min. Figure 8 depicts isocratic separations of eight pesticides within 0.7 min (~ 40 s). The application of shell particle C8 columns is not well developed; however; they has been used for ultrafast separations of molecules such as lorazeopam and virginiamycin, uracil, phenol, 4-chloro-1-nitrobenzene, and naphthalene. Acids and bases also can be resolved successfully on this column (6).

Figure 8: Chromatograms of pesticides on a shell particle column (83). Column: 50 mm à 4.6 mm Halo C18; mobile phase: 45:55 (v/v) acetonitrileâwater; flow rate: 4.0 mL/min; temperature: 60 °C; pressure: 230 bar; detection: UV absorbance at 245 nm. Peaks: 1 = tebuthiron, 2 = thiazuron, 3 = fluometuron, 4 = diuron, 5 = propanil, 6 = siduron, 7 = linuron, 8 = diflubenzuron.
If C8 and C18 phases are not good, especially for certain acidic and basic compounds, shell particle reversed-phase amide columns work well. Normally, these sort of stationary phases are quite good for highly water soluble compounds that require high aqueous mobile phases in which the polar amide phases are fully wetted. The mixtures of water and organic solvents are the best eluents, making them ideally suitable to couple with mass spectrometry (MS) detection. Most important applications of these phases are benzyl alcohol, benzylbenzoate, N,N-dimethylaniline, 2-chlorophenol, butyl paraben, 3-ethylphenol, 4-chloro-3-nitroanisole, 2-nitroaniline, 4-bromoacetanilide, 2,2'-biphenol, and benzyl benzoate. Normally, the order of retention on these phases is acids > bases > neutral molecules. HILIC silica gel columns are bare or having small organic moieties to increase wetability. Hence, a hydrophobic (mostly organic) mobile phase is used (partly aqueous eluents high in acetonitrile or sometimes methanol), resulting in an increase of retention with hydrophilic solutes (84). The application of the reversed-phase amide columns are limited but can be used efficiently if MS, inductively coupled plasma (ICP), or atomic absorption spectroscopy (AAS) are required for detection. Hemström and Irgun (85) reviewed HILIC and discussed the separation mechanism and applications. Pesek and Matyska (86) also compared the application of HILIC with other phases and found it suitable for certain compounds. Recently, McCalley (87) investigated sample capacity, column efficiency, and variation in flow rates on HILIC columns for separation of basic compounds. Efficiencies of 1,000,000 theoretical plates were reported. The separated molecules on HILIC columns are phenol, 2-naphthalenesulfonic acid, p-xylenesulfonic acid, caffeine, nortriptyline, diphenhydramine, benzylamine, and procainamide. The other applications of shell particle columns are summarized in Table II.


Table II: The Applications of superficially porous columns
Monolithic versus Shell Particle Columns
It is a difficult task to compare monolithic and shell particle columns as the applications of the latter are not as well developed. However both columns can provide analyses within few minutes, or even seconds in case of shell particle columns. Both types of columns can be used with conventional HPLC instruments. Both stationary phases can be used at moderately high pressure. The good loading capacities of these columns may also prove them to be semi-preparative in nature. We have seen applications of shell particle columns with run times of a few seconds but no monolithic column has been shown capable of providing such ultra fast analyses. Therefore, we can certainly assess that shell particle columns are faster than monolithic columns. A relative comparison of mobile phase velocity and pressures can be observed from Figures 3 and 6, respectively. And a perusal of these figures indicates that both columns are complimentary in nature. The most important observed difference is that shell particle columns are quite good for small molecules while monolithic are better for large molecules. However, few papers are available in the literature describing the separation of macromolecules also using shell particle columns. Kaczmarski and Guiochon (94) described numerical calculations of band profiles under nonlinear conditions and demonstrated the use of shell particle columns in the separations of proteins or other large molecules. Shell particle columns are available in different four types while monolithic columns are not so developed and still need more modifications and developments, especially reversed-phase modes. In addition, it is desirable to prepare monolithic phases that can produce high electroosmotic flow without effecting chemical and chromatographic properties of monolithic silica columns.
Future Perspectives and Conclusion
Certainly, both types of columns provide fast analyses and are well positioned for the future demands of the chromatography laboratory. To the best of our experience the future of shell particle and monolithic columns is quite good for several reasons. The working capacities and efficiencies of these phases make them ideal for a wide range of molecules, and eventually they must also be available as chiral columns due to great demand of enantioseparations in drug development and design. As discussed previously, monolithic columns have been in use for several years but shell particle columns are relatively new. Most probably, there will be a tight competition of monolithics with shell particle columns in the near future. Of course, the future will decide the fate of these fast columns but our experience dictates that shell particle columns may deserve a better reputation in ultrafast separation science.
Acknowledgment
One of the authors (IA) is thankful to Professor Mohammed S. Khan, Head, Department of Chemistry, Sultan Qaboos University for his invitation to be a Visiting Professor to the Department of Chemistry.
Imran Ali*, Fakhr Eldin O.Suliman*, and Hassan Y. Aboul-Enein†
*Department of Chemistry, College of Sciences Sultan Qaboos University, Muscat, Oman
Please direct correspondence to drimran_ali@yahoo.com
References
(1) J.N. Done, G.J. Kennedy and J.H. Knox, Nature 237, 77–81 (1972).
(2) R.E. Majors, LCGC 24(3), 248–266 (2006).
(3) J.W. Thompson, J.S. Mellors, J.W. Eschelbach, and J.W. Jorgenson, LCGC 24(1), 16–21 (2006).
(4) J.J. Kirkland, J. Chromatogr. Sci. 38, 535–544 (2000).
(5) Chromlith R Prep: Increase in speed, efficiency and productivity, Merck KGaA, 64271 Darmstadt, Germany, www.chemdat.info.
(6) Guide to ultra fast HPLC, Quick tips for converting conventional reversed phase HPLC separations to ultra fast separations, MAC-MOD Analytical, Inc. 103 Commons Court, Chadds Ford, PA 19317, USA, 2007, www.mac-mod.com.
(7) F. Svec, T.B. Tennikova, and Z. Deyl, Monolithic Materials: Preparation, Properties, and Applications (Elsevier, Amsterdam, 2003), pp. 19–50.
(8) K. Cabrera, D. Lubda, H.M. Eggenweiler, H. Minakuchi and K. Nakanishi, J. High. Resolut. Chromatogr. 23, 93–99 (2000).
(9) D. Josic, A. Buchacher, and A. Jungbauer, J. Chromatogr., B 752, 191–205 (2001).
(10) N. Tanaka, H. Kobayashi, N. Ishizuka, H. Minakuchi, K. Nakanishi, K. Hosoya and T. Ikegami, J. Chromatogr., A 965, 35-49 (2002).
(11) AM. Siouffi, J. Chromatogr., A 1000, 801–818 (2003).
(12) K. Cabrera, J. Sep. Sci. 27, 843–852 (2004).
(13) A. Podgornik and A. Strancar, Biotechnol. Ann. Rev. 11, 281–333 (2005).
(14) F. Svec. J. Sep. Sci. 28, 729–745 (2005).
(15) F. Svec and C.G. Huber, Anal. Chem. 78, 2100–2107 (2006).
(16) H. Kobayashi, T. Ikegami, H. Kimura, T. Hara, D. Tokuda, and N. Tanaka, Anal. Sci. 22, 491–501 (2006).
(17) G. Guiochon. J. Chromatogr., A 1168, 101–168 (2007).
(18) M. Barut, A. Podgornik, L. Urbas, B. Gabor, P. Brne, J. Vidic, S. Plevcak, and A. Strancar, J. Sep. Sci. 31, 1867–1880 (2008).
(19) J. Urban and P. Jandera, J. Sep. Sci. 31, 2521–2540 (2008).
(20) T. Ikegami, K. Tomomatsu, H. Takubo, K. Horie, and N. Tanaka, J. Chromatogr., A 1184, 474–503 (2008).
(21) O. Nunez, K. Nakanishi, and N. Tanaka, J. Chromatogr., A 1191, 231–252 (2008).
(22) R. Wu, L. Hu, F. Wang, M.Ye, and H. Zou, J. Chromatogr., A 1184, 369–392 (2008).
(23) H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka, and N. Tanaka, J. Chromatogr., A 762, 135–146 (1997).
(24) H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka, and N. Tanaka, J. Chromatogr., A 797, 121–131 (1998).
(25) D. Josic and A. Strancar, Ind. Eng. Chem. Res. 38, 333–342 (1999).
(26) F. Svec and J.M.J. Frechet, Science 273, 205–211 (1996).
(27) N. Tanaka, H. Nagayama, H. Kobayashi, T. Ikegumi, K. Hosoya, N. Ishizuka, H. Minakuchi, K. Nakanishi, K. Cabrera, and D. Lubda, J. High Resolut. Chromatogr. 23, 111 (2000).
(28) N. Ishizuka, H. Minakuchi, K. Nakanishi, N. Soga, H. Nagayama, K. Hosoya, and N. Tanaka, Anal. Chem. 72, 1275–1280 (2000).
(29) E. Sugrue, P. Nesterenko, and B. Paull, J. Sep. Sci. 27, 921–930 (2004).
(30) D. Lubda, W. Lindner, M. Quaglia, C. du Fresne von Hohenesche, and K.K. Unger, J. Chromatogr., A 1083, 14–22 (2005).
(31) K. Bencina, M. Bencina, A. Podgornik, and A. Tramcar, J. Chromatogr., A 1160, 176–183 (2007).
(32) N. Vervoort, P. Gzil, G.V. Baron, and G. Desmet, Anal. Chem. 75, 843–850 (2003).
(33) P. Persson, O. Baybak, F. Plieva, I.Y. Galaev, B. Mattiasson, B. Nilsson, and A. Axelsson, Biotechnol. Bioeng. 88, 224–236 (2004).
(34) N. Vervoort, P. Gzil, G.V. Baron, and G. Desmet, J. Chromatogr., A 1030, 177–186 (2004).
(35) J. Billen, P. Gzil, and G. Desmet, Anal. Chem. 78, 6191–6201 (2006).
(36) P. Gzil, J. De Smet, and G. Desmet, J. Sep. Sci. 29, 1675–1685 (2006).
(37) P. Gzil, N. Vervoort, G.V. Baron, and G. Desmet, J. Sep. Sci. 27, 887–896 (2004).
(38) G. Desmet, D. Cabooter, P. Gzil, H. Verelst, D. Mangelings, Y. Vander Heyden, and D. Clicq, J. Chromatogr., A 1130, 158–166 (2006).
(39) H.Y. Aboul-Enein, I. Ali and Hubert Hoenen, Biomed. Chromatogr. 20, 760–764 (2006).
(40) I. Ali and H.Y. Aboul-Enein, J. Liq. Chromatogr. & Relat. Technol. 28, 3169–3179 (2005).
(41) H.Y. Aboul-Enein and I. Ali, Talanta 65, 276–280 (2004).
(42) I. Ali and H.Y. Aboul-Enein, Anal. Lett. 37, 2351–2361 (2004).
(43) I. Ali, V.K. Gupta, Prashant Singh, H.V. Pant, and H.Y. Aboul-Enein, J. Liq. Chromatogr. & Relt. Technol. 31, 2862–2878 (2008).
(44) T. Hara, H. Kobayashi, T. Ikegami, K. Nakanishi, and N. Tanaka, Anal. Chem. 78, 7632–7642 (2006).
(45) K.D. Bartle and P. Myers, J. Chromatogr., A 916, 3–23 (2001).
(46) G. Puy, C. Demesmay, J.L. Rocca, J. Iapichella, A. Galarneau and D. Brunel, Electrophoresis 27, 3971–3980 (2006).
(47) M.C. Breadmore, S. Shrinivasan, K.A. Wolfe, M.E. Power, J.P. Ferrance, B. Hosticka, P.M. Norris, and J.P. Landers, Electrophoresis 23, 3487–3495 (2002).
(48) C. Gatschelhofer, M.G. Schmid, K. Schreiner, T.R. Pieber, F.M. Sinner, and G. Gübitz, J. Biochem. Biophys. Methods 69, 67–77 (2006).
(49) B. Chankvetadze, C. Yamamoto, N. Tanaka, K. Nakanishi and Y. Okamoto, J. Sep. Sci. 27, 905–911 (2004).
(50) B. Chankvetadze, T. Kubota, T. Ikai, C. Yamamoto, M. Kamigaito, N. Tanaka, K. Nakanishi, and Y. Okamoto, J. Sep. Sci. 29, 1988–1995 (2006).
(51) N. Ishizuka, H. Kobayashi, H. Minakuchi, K. Nakanishi, K. Hirao, K. Hosoya, T. Ikegami, and N. Nakanishi, J. Chromatogr., A 960, 85–96 (2002).
(52) S. Murko, R. Milacic, and J. Scancar, J. Inorg. Biochem. 101, 1234–1241 (2007).
(53) F. Smrekar, M. Ciringer, M. Peterka, A. Podgornik, and A. Tramcar, J. Chromatogr., B 861, 177–180 (2008).
(54) P.T. Vallano, R.S. Mazenko, E.J. Woolf, and B.K. Matuszewski, J. Chromatogr., B 779, 249–257 (2002).
(55) S. Ota, S. Miyazaki, H. Matsuoka, K. Morisato, Y. Shintani, and K. Nakanishi, J. Biochem. Biophys. Methods 70, 57–62 (2007).
(56) M. Brgles, B. Halassy, J. Tomasic, M. Santak, D. Forcic, M. Barut, and A. Strancar, J. Chromatogr., A 1144, 150–154 (2007).
(57) D. Forcic, K. Branovic-Cakanic, J. Ivancic, R. Jug, M. Barut, and A. Strancar, J. Chromatogr., A 1065, 115–120 (2005).
(58) S. Jerman, A. Podgornik, K. Cankar, N. Cadet, M. Skrt, J. Zel, and P. Raspor, J. Chromatogr., A 1065, 107–113 (2005).
(59) H. Minakuchi, N. Ishizuka, K. Nakanishi, N. Soga, and N. Tanaka, J. Chromatogr., A 828, 83–90 (1998).
(60) Q. Xu, M. Mori, K. Tanaka, M. Ikedo, W. Hu, and P.R. Haddad, J. Chromatogr., A 1041, 95–99 (2004).
(61) K. Branovic, G. Lattner, M. Barut, A. Strancar, D.J. Josic, and A. Buchacher, J. Immunol. Methods 271, 47–58 (2002).
(62) P. Brne, A. Podgornik, K. Bencina, B. Gabor, A. Strancar, and M. Peterka, J. Chromatogr., A 1144, 120–125 (2007).
(63) M. Rucevic, J.G. Clifton, F. Huang, X. Li, H. Callanan, D.C. Hixson, and D. Josic, J. Chromatogr., A 1123, 199–204 (2006).
(64) K. Isobe and Y. Kawakami, J. Chromatogr., A 1065, 129–134 (2005).
(65) A.M. van Nederkassel, A. Aerts, A. Dierick, D.L. Massart, and Y.V. Heyden, J. Pharm. & Biomed. Anal. 32, 233–249 (2003).
(66) P.A. Sutton and P.N. Nesterenko, J. Sep. Sci. 30, 2900–2909 (2007).
(67) I. Vovk and B. Simonovska, J. Chromatogr., B. Anal. Technol. Biomed. Life Sci. 849, 337–343 (2007).
(68) I. Vovk, B. Simonovska, and M. Bencina, J. Chromatogr., A 1065, 121–128 (2005).
(69) H.M. Oliveiraa, M.A. Segundoa, J.L.F.C. Limaa, and V. Cerdà Talanta 77, 1466–1472 (2009).
(70) P. Kramberger, N. Petrovic, A. Strancar, and M. Ravnikar, J. Virol. Methods 120, 51–57 (2004).
(71) S.A. Bellomarinoa, A.J. Brown, X.A. Conlana, and N.W. Barnett, Talanta 77, 1873–1876 (2009).
(72) I. Vovk and B. Simonovska, J. Chromatogr., A 1144, 90–96 (2007).
(73) M. Peterka, M. Jarc, M. Banjac, V. Frankovic, K. Bencina, M. Merhar, V. Gaberc-Porekar, V. Menart, A. Strancar, and A. Podgornik, J. Chromatogr., A 1109, 80–85 (2006).
(74) D.V. McCalley, J. Chromatogr., A 965, 51–64 (2002).
(75) J. Li, G. Wu, and Y. Zhu, J. Chromatogr., A 1118, 151–154 (2006).
(76) S. Murko, R. Milacic, and J. Scancar, J. Inorg. Biochem. 101, 1234–1241 (2007).
(77) Y. Tian, C. Zhong, E. Fu, and Z. Zeng, J. Chomatogr., A 1216, 1000–1007 (2009).
(78) J.J. Kirkland, F.A. Truszkowski, C.H. Dilks, and G.S. Engel, J. Chromatogr., A 890, 3–13 (2000).
(79) J.J. Kirkland, T.J. Langlois, and J.J. DeStefano, Am. Lab. 39, 18–21 (2007).
(80) J.M. Cunliffe and T.D. Maloney, J. Sep. Sci. 30, 3104–3109 (2007).
(81) F. Gritti, A. Cavazzini, N. Marchetti, and G. Guiochon, J. Chromatogr., A 1157, 289–303 (2007).
(82) F. Gritti and G. Guiochon, J. Chromatogr., A 1166, 30–46 (2007).
(83) J.J. DeStefano and T.J. Langlois, J. Chromatogr. Sci. 46, 254–260 (2008).
(84) A.J. Alpert, J. Chromatogr., A 499, 177–196 (1990).
(85) P. Hemström and K. Irgun, J. Sep. Sci. 29, 1784–1821(2006).
(86) J. Pesek and M.T. Matyska, LCGC 25(5), 480–490 (2007).
(87) D.V. McCalley, J. Chromatogr., A 1193, 85–91 (2008).
(88) A. Cavazzini, F. Gritti, K. Kaczmarski, N. Marchetti, and G. Guiochon, Anal. Chem. 79, 5972–5979 (2007).
(89) F. Gritti and G. Guiochon, J. Chromatogr., A 1176, 107–122 (2007).
(90) N. Marchetti and G. Guiochon, J. Chromatogr., A 1176, 206–216 (2007).
(91) N. Marchetti, A. Cavazzini, F. Gritti, and G. Guiochon, J. Chromatogr., A 1163, 203–211 (2007).
(92) F. Gritti and G. Guiochon, J. Chromatogr., A 1216, 63–78 (2009).
(93) Z. Bayram-Hahn, B.A. Grimes, A.M. Lind, R. Skudas, K.K. Unger, A. Galarneau, J. Iapichella, and F. Fajula, J. Sep. Sci. 30, 3089–3103 (2007).
(94) K. Kaczmarski and G. Guiochon, Anal. Chem. 79, 4648–4656 (2007).




















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

