Sulfated β-Cyclodextrin as Chiral Mobile Phase Additive for Ultrahigh-Pressure Liquid Chromatography
Special Issues
A fast enantiomeric separation of a chiral aromatic amine was achieved, using ultra high pressure liquid chromatography and highly sulfated β-cyclodextrin (S-β-CD) as a chiral additive in the mobile phase. The stationary phase consisted of a core shell support with a particle size of 2.7 µm. Under these conditions the baseline separation was obtained within 2.5 min. The influence of the concentration of the additive, along with the thermodynamics of the separation, were studied. Molecular mechanics calculations were consistent with the experimental data for the order of elution, providing further evidence of these interactions. The enantiomeric separation at high temperature (90 °C) using only water as mobile phase also was achieved for the first time.
Separation of enantiomers using chiral mobile phases is a technique that was used widely in the 1980s and early 1990s, due to the lack of chiral stationary phases. The technique has several advantages compared to chiral stationary phases. For instance, it employs achiral stationary phases, which are less expensive and more rugged than a chiral stationary phase. At the same time it allows more flexibility because a column can be used in conjunction with several additives at the same time or the chiral additive can be eluted out of the column with a strong solvent and a subsequent additive can be used. The disadvantage of the method stems from the existence of a secondary chemical equilibrium, which has a great contribution to the band broadening of the two enantiomeric analytes and makes the mechanism of separation difficult to understand.


The introduction of core shell technology particles of 2.7 μm particle size, along with a porous shell of 0.5 μm (1–4), revitalized chiral separation with chiral mobile phases. Fast kinetics of mass transfer, along with a fast distribution of the analyte–complex, minimizes the secondary chemical equilibrium and consequently brings substantial improvement to the band broadening. Such stationary phases, along with ultrahigh-pressure liquid chromatography (UHPLC), bring improved timeliness and efficiency to chiral separations with chiral mobile phases.
Sulfated β-CD (S-β-CD) represents a class of anionic derivatives of CDs, and has been used extensively in capillary electrophoresis (5,6). Stalcup and colleagues (7) used this selector as a chiral stationary phase to enantioresolve 33 compounds, among which six did not have an amine functional group. There are very few examples of the use of S-β-CD as an additive in chiral mobile phases. For instance, Ameyibor and colleagues (8) used S-β-CD as an additive in the mobile phase for the enantiomeric separation of some selected chiral drugs.
In this article, we present the enantiomeric separation of an amine using core shell technology with sulfate β-CD as the chiral mobile phase selector and UHPLC. We first assess the influence of the concentration of the additive and the thermodynamics of the separation on chromatographic conditions. We further explore the molecular interactions between enantiomers and S-β-CD using VCD spectroscopy as well as molecular modeling.
Experimental
The chromatographic experiments were performed on a Waters Acquity UPLC system equipped with a vacuum degasser, a high pressure binary pump, a column oven, and a photodiode-array detector (Waters, Milford, Massachusetts). The separations were performed on two 150 mm × 4.6 mm Halo columns (Halo C8 and Halo C18, Advanced Material Technology, Wilmington, Delaware) with 2.7-μm particle size. The highly sulfated β-CD, acetonitrile and water were purchased from Sigma Aldrich (Milwaukee, Wisconsin). The mobile phase consisted of a solvent mixture of 15% acetonitrile in water containing different amounts of S-β-CD, depending upon the experiment. The mobile phase was premixed before the chromatography and was pumped through the system at a flow rate of 1.5 mL/min, which generated a back pressure of ~11,000 psi.
The column was equilibrated with the mobile phase until a steady baseline was obtained. The column void time, to, was determined by measuring the first baseline perturbation. Each experiment was performed three times at each mobile phase composition and temperature change. The chromatography at high temperature was performed on a Waters X-bridge C8 column (75 mm × 4.6 mm, 3-μm particle size). The temperature was maintained at a constant 90 °C using a Selerity high temperature oven (Salt Lake City, Uah) connected to a binary pump 1100 Agilent HPLC system (Agilent Technologies, Santa Clara, California). The two enantiomers of the chiral amine hydrochloride salt of compound 1 used in this study were synthesized in house. The retention factor, k', was obtained using: k'=(tR - to)/to, where tR is the retention time of the analyte. The selectivity factor, α, was calculated as α = k2'/k1', where k1' and k2' are the retention factors of the first and second eluted enantiomer, respectively.
All computations on S-β-CD and its complexes with each enantiomer were performed using Hyperchem software (Hypercube Inc., Gainesville, Florida). Structure optimizations and energy minimizations were implemented using a molecular mechanics force field, MM+ method. Docking of the enantiomers was done initially to maximize the electrostatic interaction and hydrophobic interaction between the enantiomer and S-β-CD.
Results and Discussion
The structure of the compound under study is presented in Figure 1. The compound has a tertiary amine and an aromatic moiety. The tertiary amine group has three ethyl groups attached. It is expected that steric hindrance will be a major factor impeding interaction at these sites. Because the structure of the compound shows few sites of interaction, we pursued the separation using a porous shell C8 and C18 reversed-phase stationary phase along with a mobile phase containing S-β-CD as an additive. The tertiary amine has a pKa of ~10.8 and is expected to be protonated at lower pHs. Under these conditions the interactions with the S-β-CD can be assumed as both electrostatic interactions between the amine and the sulfate groups of cyclodextrins, and inclusion inside the cavity of S-β-CD.

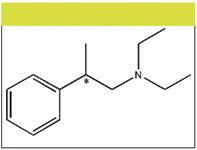
Figure 1: Structure of Compound 1. The chiral center is labeled with an asterisk (*).
Influence of concentration of sulfated β-CD on the separation: The presence of cyclodextrin as a chiral additive in the mobile phase induces a secondary chemical equilibrium (Scheme 1) (9,10).

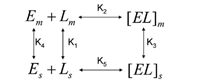
where subscript s and m refer to the species present in the stationary phase and in the mobile phase respectively, and K1, K2, K3, K4, and K5 are the equilibrium constants for each process.
Under the same mobile phase conditions used for the separation of the two enantiomers of compound 1, S-β-CD exhibits nearly no retention (elutes out close to the dead volume) on both the C8 and C18 stationary phases. This indicates that the adsorption of this chiral additive on these stationary phases is minimal and the complexation with the chiral additive occurs mainly in the mobile phase.
To assess the influence of the chiral additive on the separation, the concentration of S-β-CD was varied in the mobile phase from zero to 2.0 mM. An example of baseline separation under 2.5 min is presented in Figure 2.

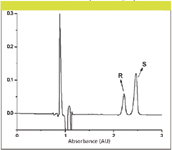
Figure 2: Separation of compound 1 on UHPLC. Column: 150 mm à 4.6 mm, 2.7-μm Halo C8; mobile phase: 4 g/L S-β-CD in 88:12 (v/v) waterâacetonitrile; flow rate: 1.6 mL/min; temperature: 30 °C
The influence of the concentration of the additive on the retention factor (k') of the two enantiomers, and on the selectivity, α, is presented in Figure 3.

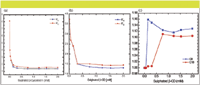
Figure 3: Influence of the concentration of S-β-CD on retention factor: (a) Halo C8, (b) Halo C18, and (c) selectivity factor.
The data of Figure 3 indicate that, upon increasing the concentration of the additive in the mobile phase, the retention factor decreases drastically from zero to 0.25 mM then remains constant for the rest of the range of the concentrations used in the study. Such results indicate that the complexation between the sulfated β-CD and each enantiomer occurs in the mobile phase. The enantioselectivity, α, increases with the increase in concentration of the chiral additive, as shown in Figure 3c. Interestingly, the enantioselectivity is higher on the C8 stationary phase compared to the C18 stationary phase. Fourier-transform attenuated total reflection infrared (FT-ATR-IR) spectroscopy of the two stationary phases showed that these results are due to the difference in ligand density of the C8 versus the C18 on the silica support, which is smaller on the C18 compared with the C8 (10). As a consequence there is a possibility that there might be residual silanols that can produce an achiral interaction that impedes the enantioselectivity on the C18 stationary phase.
Influence of temperature on separation: For an isobaric, isothermal chromatographic process, the effect of temperature on the capacity factor can be expressed by the van't Hoff equation (11):

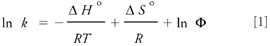
where ΔΔHo and ΔΔSo are the standard enthalpy and entropy of solute transfer from mobile phase to the stationary phase, and Φ is the phase ratio. According to equation 1, a plot of ln k' vs. 1/T should yield a straight line with a slope of –ΔHo/R and an intercept of (ΔSo/R + ln Φ). Deviation from linearity can be indicative of a conformational change of the chiral phase (12–14).
The separation factor for a given pair of enantiomers (α = k'2/k'1) is a measure of the enantioselectivity and represents the difference in the free energy of the interactions of the two enantiomers with the chiral phase.

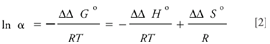
A plot of ln α vs. 1/T will yield a straight line with a slope of – ΔΔHo/R and an intercept of ΔΔSo/R, providing that the enantioselective interactions do not change over the temperature range of the study.
In our study, the temperature was varied between 10 °C and 60 °C. Plots of ln k' vs. 1/T for the separation of the two enantiomers of compound 1 on both stationary phases, C8 and C18, are presented in Figures 4a and 4b.

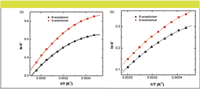
Figure 4: van Hoff plots in k for the two enantiomers: (a) Halo C 8 column; (b) Halo C18 column.
Both plots of Figures 4a and 4b indicated that the relationship between ln k' and temperature is nonlinear. Plots of ln α vs. 1/T are presented in Figures 5a and 5b. The plot generated from the chromatographic data on the C18 stationary phase shows a linear behavior (r2 > 0.999), indicating that temperature has the same effect on both enantiomers (Figure 5b). The plot generated from the chromatographic data on the C8 stationary phase shows nonlinear behavior; however it was possible to divide the plot into two linear regions with a change in slope at ~40 °C (Figure 5a). Such behavior indicates a change in conformation of the chiral additive (10).

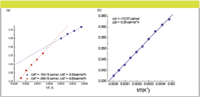
Figure 5: van Hoff plot in selectivity for (a) Halo C8 and (b) Halo C18 columns.
Interestingly, the values for ΔΔHo and ΔΔSo in the high temperature region are almost twice those at lower temperature (Figure 5). Such results can be interpreted as follows: at lower temperatures both the cyclodextrin and the enantiomeric analytes are solvated. As the temperature increases, a thermal desolvation of the sulfated β-CD occurs. As a consequence the hydrophobic interaction between the two enantiomers and the interior of the cyclodextrin cavity, as well as the electrostatic interaction between the tertiary amine of the enantiomers and the SO3- , are enhanced. Thus, the ΔΔHo becomes more negative and the interaction becomes entropically unfavorable. At the same time, as ΔΔHo becomes more negative, ΔΔSo also becomes more negative in the high temperature region (compared to the lower temperature region), as a result of enthalpy–entropy compensation. The fact that ΔΔHo is higher in the higher temperature region relative to lower temperature region suggests that one can obtain separation of the two enantiomers employing higher temperature chromatography. Indeed, Figure 6 shows the enantiomeric separation of compound 1 at 90 °C.

Figure 6: Separation of compound 1 using high temperature. Column: 75 mm à 4.6 mm, 3-μm Waters Xbridge C8; mobile phase: 8 g/L S-β-CD in water; flow rate: 0.7 mL/min; temperature: 90 °C.
Molecular modeling of the complex of S-β-CD with each enantiomer of compound 1: The interactions between S-β-CD and each enantiomer were further assessed using molecular modeling. Based upon previous vibrational circular dichroism and infrared spectroscopy studies (10), each enantiomer of compound 1 was docked with S-β-CD such that the electrostatic interactions occurred between each enantiomer and S-β-CD through the protonated nitrogen of the compound and the negatively charged –SO3- groups of S-β-CD located at the narrow part of the annuli. At the same time an inclusion occurred through the hydrophobic interaction with the phenyl group and the cavity of S-β-CD. In Figures 7a and 7b, we present the optimized structure of the two complexes of each enantiomer with S-β-CD. It can be observed that the nitrogen of each enantiomer is structurally close to the sulfate groups, at a distance of 3.58 Å and 3.63 Å for the R- and S-enantiomers, respectively. In addition, the phenyl groups of both enantiomers are included in the cavity of S-β-CD. The R-enantiomer is slightly closer to the interior wall of the annulus than the S-enantiomer. These results suggest that the interaction between the R-enantiomer and the S-β-CD is stronger than for the S-enantiomer.

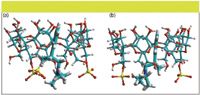
Figure 7: Optimized structure of (a) S-β-CD complex with R-enantiomer and (b) S-β-CD complex with S-enantiomer.
The free energy obtained from molecular simulation shows that the R-enantiomer has a greater potential interaction with S-β-CD relative to the S-enantiomer. The energy values are 170.89 kcal/mol and 171.63 kcal/mol (1 cal = 4.18 J), for the complex of S-β-CD with the R-enantiomer and the S-enantiomer, respectively. These values are consistent with the elution order observed in the chromatographic experiment, which shows that the R-enantiomer interacts strongly with S-β-CD and consequently is eluted first. The difference in the interaction energies between the two enantiomers (E = 0.74 kcal/mol), represents the energetic contribution to enantioselectivity. Although the absolute energy difference does not correlate well with that obtained from the chromatographic experiments, the elution order is consistent with the relative energies from the theoretical calculation.
Conclusion
In conclusion, this report demonstrates the chiral separation of a chiral amine compound using S-β-CD as a chiral additive in the mobile phase. It was found that S-β-CD undergoes a conformational change with increasing temperature. The interaction between the between the studied compound an S-β-CD occurs through inclusion by hydrophobic interactions with the interior of the macrocycle. In addition, electrostatic interactions occur between the tertiary amine portion of the molecule and the sulfated groups located at the narrow part of the rim of the sulfated cyclodextrin. The values for Ho at higher temperature region led to the achievement of enantiomeric separation at high temperature.
Acknowledgments
We thank Dr. Joseph J. Destefano from Advanced Materials Technology, Inc. (Wilmington, Delaware) for supplying the Halo C18 and C8 stationary phases.
Shengli Ma, Sherry Shen, Nizar Haddad, Wenjun Tang, Jing Wang, Heewon Lee, Nathan Yee, Chris Senanayake, and Nelu Grinberg
Development Department, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut.
Please direct correspondence to nelu.grinberg@boehringer-ingelheim.com
References
(1) F. Gritti, A. Cavazzini, N. Marchetti, and G. Guiochon, J. Chromatogr., A 1157, 189 (2007).
(2) F. Gritti and G. Guiochon, J. Chromatogr., A 1176, 107 (2007).
(3) F. Gritti and G. Guiochon, J. Chromatogr., A 1169, 125 (2007).
(4) N. Marchetti and G. Guiochon, J. Chromatogr., A 1176, 206 (2007).
(5) C. E. Evans and A.M. Stalcup, Chirality 15, 709 (2003).
(6) S.R. Gratz and A.M. Stalcup, Anal. Chem. 70, 5166 (1998).
(7) A.M. Stalcup and K.H. Gahm, Anal. Chem. 68, 1369 (1996).
(8) E. Ameyibor and J.T. Stewart, J. Liq. Chrom. & Rel. Technol 20, 855 (1997).
(9) N. Grinberg, T. Burakowski, A.M. Stalcup, in HPLC for Pharmaceutical Scientists, Y. Kazakevich and R. LoBrutto, Eds. (Wiley-Interscience, New York, 2007), p. 987.
(10) S. Ma, S. Shen, N. Haddad, W. Tang, J. Wang, H. Lee, N. Yee, C. Senanayake, and N. Grinberg, J. Chromatogr., A. 1216, 1232 (2009).
(11) Cs. Horvath, W.R. Melander, in Fundamentals and Applications of Chromatographic and Electrophoretic Methods, E. Heftman, Ed. (Elsevier Scientific Publishing Company, Amsterdam, 1983), p. A26.
(12) M. See, D.R. Sidler, A.J. Simon, C.R. Middaugh, R. Thompson, L.J. August, H.J. Perpall, and N. Grinberg, Chirality 11, 224 (1999).
(13) T.O'Brien, L. Crocker, R. Thompson, K. Thompson, P.H. Toma, D. A. Conlon, B. Feibush, C. Moeder, G. Bicker, and N. Grinberg, Anal. Chem. 69, 1999 (1997).
(14) Y. Bereznitski, R. LoBrutto, R. Thompson, K. Thompson, P. Sajonz, L. S. Crocker, J. Kowal, D. Cai, M. Journet, T. Wang, J. Wyvratt, and N. Grinberg, Enantiomer 7, 305 (2002).















Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.

Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

