Split Injection GC: Setting the Split Ratio in Shoot-and-Dilute GC
Jack Cochran’s new column “Practical GC” provides readers with practical advice and new experimental evidence for how to get the best results from their gas chromatography (GC) systems. This instalment looks at understanding and using split ratio for “shoot and dilute” GC.
Jack Cochran, Restek Corporation, Bellefonte, Pennsylvania, USA.


Photo Credit: Liam Larkin/ EyeEm/ Getty Images

Jack Cochran’s new column “Practical GC” provides readers with practical advice and new experimental evidence for how to get the best results from their gas chromatography (GC) systems. This instalment looks at understanding and using split ratio for “shoot and dilute” GC.
For my introductory “Practical GC” article on split injection gas chromatography (GC), I listed the advantages of this technique and showed recently collected experimental evidence for how split injection allows users to avoid sensitive compound degradation, to successfully use polar solvents with non-polar GC columns, and to start at higher GC oven temperatures to increase throughput.1 I also suggested some basic conditions for using “shoot-and-dilute” GC to get these advantages, most notably a precision-type liner for accurate sample transfer and homogenization, and a starting split ratio of 10 or more. The second article in the series used comparisons against other liners to help cement the precision-type liner as the split liner of choice.2 It’s time to focus on split ratio, which is mainly used to control the amount of sample introduced to the GC column.
When split injection for dirty environmental or food samples is used in residue analysis to help keep systems up longer, the obligation is to introduce enough sample to the GC column, and finally, the detector, such that desired limits of quantification can be reliably achieved. That makes a split ratio of 1:10 (hereinafter referred to as split ratio of 10) a good starting point because the higher flow through the inlet versus splitless injection suppresses compound degradation and adsorption on liner surfaces. By definition, a split ratio of 10 means that one part of the injected sample goes to the GC column and 10 parts go out the split vent, so ~10x less problem-causing non-volatile material is delivered to the GC column with each injection. Tienstra et al. recently used a split ratio of 10 to great advantage for the analysis of pesticides and other contaminants in animal feed QuEChERS extracts by GC paired with ultra-sensitive atmospheric pressure chemical ionization tandem mass spectrometry (APCI–MS–MS).3 Some highlights from their paper included:
1. Fast GC run times of less than 10 min and fast cycle times because of higher initial oven start temperatures allowed by using split injection.
2. Less co-extracted matrix on the GC column and in the MS source leading to greater uptime because of split injection.
3. Better GC stationary phase compatibility with QuEChERS acetonitrile extracts because most of the solvent does not make it to the GC column as a result of split injection.
4. Applicable to complex feed matrices with relevant limits of quantification because of the sensitive detector used.
But what about those cases where we have neat samples for analysis, or relatively high concentrations of target analytes in a solvent, or perhaps a mix of high and low concentrations of analytes of interest? How do we set our split ratio? By paying attention to what chromatograms tell us, as my colleague Jaap de Zeeuw likes to say. Setting the split ratio for non-trace-level work is the subject for this instalment of “Practical GC”.
Experimental
A 30 m x 0.25 mm, 0.25-µm Stabilwax GC column (Restek) with helium carrier gas at a constant flow of 2.0 mL/min was operated with the following GC oven program: 80 °C (0.1 min), 10 °C/min to 240 °C (2.9 min). A flame ionization detector (FID) (Agilent) at 260 °C was used with nitrogen makeup + column flow at 50 mL/min. One µL fast autosampler injections of standards containing fragrance allergens at 400 ng/µL each compound were made into a 4-mm precision-type split inlet liner (see chromatograms) at 200 °C. The split ratios used were 10, 20, 50, 100, 200, and 400. A men’s cologne was also analyzed under the same conditions.
Results and Discussion
Analyzing a standard mixture where the concentrations for each compound are the same makes it relatively easy to dial in the proper split ratio with just a few GC runs. When using 0.25 mm x 0.25 µm GC columns, I like to start with split ratios of 10, 20, 50, 100, 200, and 400 and then inspect the resulting chromatograms for good peak shapes and adequate detection of analytes. Figure 1 illustrates what I mean about good peak shapes where the higher split ratio (less compound amount on column) analyses show peaks with asymmetry values of 1.00, a value that indicates a perfectly Gaussian peak. Peaks at split ratios of 10 and 20 (more compound amount on column) are fronting and have asymmetry values of less than 1.00, an indication of column overload – something to be avoided to preserve separation efficiency and establish repeatable retention times. Figure 2 shows how retention time errors can be exaggerated when GC column overload occurs as a result of poor choice of split ratio, and also demonstrates that by controlling split ratio to introduce 8 ng or less of each compound to the 0.25 mm x 0.25 µm GC column chosen for this work, peak shapes are good, separations are better, and retention times are consistent.

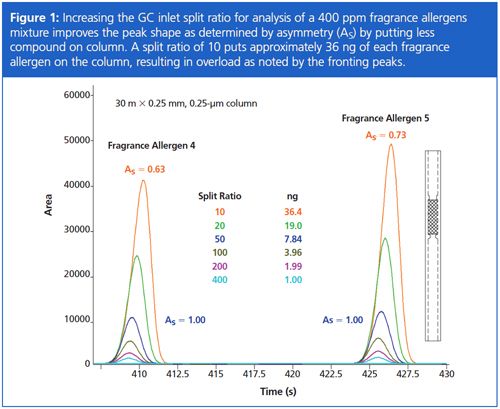

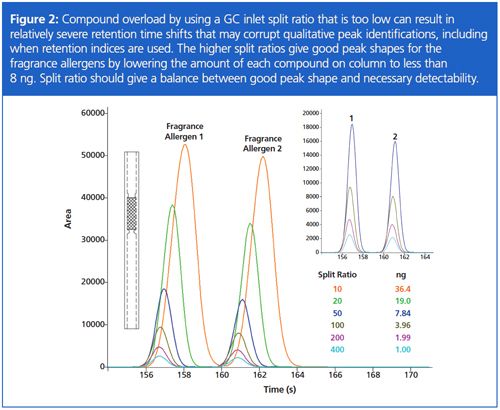
I’ve been asked by colleagues about the accuracy of split ratio, that is, as regards the expected amount of compound delivered to the GC column. Using modern electronic pneumatic control (EPC) GC inlets and a precision-type split liner that contains wool for syringe needle wipe during injection and good sample homogenization, I’ve seen excellent accuracy (and repeatability). The ability to deliver an accurately scaled amount of compound to the GC column across a range of split ratios is seen in Figure 3. From a practical standpoint this means split ratio can be chosen to deliver the desired amount of compound to the GC column without fear of discrimination. As you review Figure 3, remember too that a 400 ppm compound injected at 1 µL with a split ratio of 10 does not place 40 ng on column (1/10), but instead yields 36.3 ng – one part to GC column, 10 parts to split vent (1/11 of 400 ng is 36.4 ng).
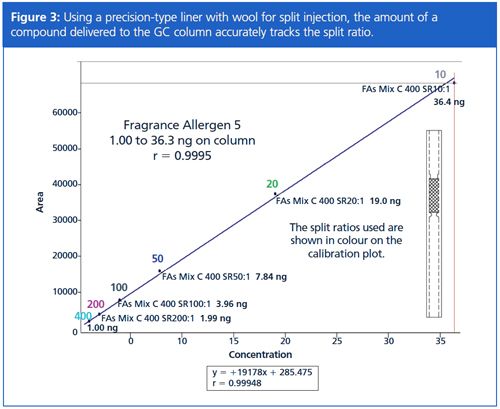
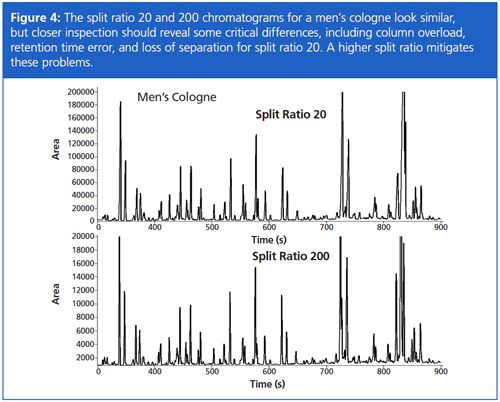
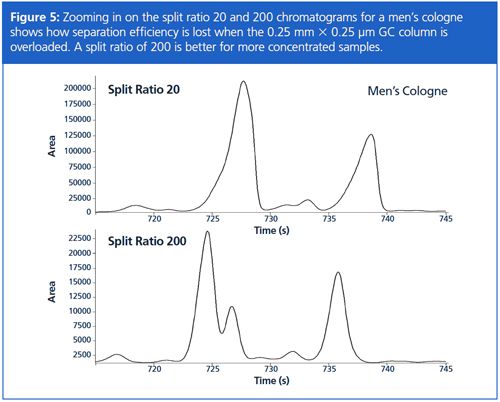
Being confident in the accuracy for a wide range of split ratios is important when analyzing samples that show extreme concentration differences, such as is seen when analyzing fragrances. Setting the split ratio has to take into consideration: (1) minimizing GC column overload; (2) detecting lower level peaks of interest; (3) maximizing separation power of the GC column; and (4) retention time repeatability. The split ratio chromatograms of 20 and 200 for a men’s cologne in Figure 4 are similar, so chromatographers might logically think to use a split ratio of 20 to improve peak detection. First though, note that generally speaking the chromatograms appear to have the same number of peaks, even though the split ratios are different by an order-of-magnitude. Closer inspection of the split ratio 20 chromatogram indicates severe overload of the GC column by some components, even to the point of causing peak coelutions (Figure 5). In this case, a split ratio of 200 is more appropriate.

Conclusions
With an appropriately chosen split liner, a few simple laboratory experiments, and some time set aside to “read” the chromatograms, a user can easily set the split ratio for good shoot-and-dilute GC work on more concentrated samples as shown in this instalment of “Practical GC”. Proper choice of GC inlet split ratio will improve chromatographic separation and retention time repeatability by avoiding column overload, while still allowing adequate detection for all the compounds of interest.
References
[1] Jack Cochran, The Column11(21), 14–20 (2015).
http://images2.advanstar.com/PixelMags/lctc/digitaledition/November23-2015-uk.html#14
[2] Jack Cochran, The Column12(4), 10–15 (2016).
http://images2.advanstar.com/PixelMags/lctc/digitaledition/March09-2016-uk.html#10
[3] M. Tienstra, T. Portolés, F. Hernández, and J.G. Mol, Journal of Chromatography A1422, 289 (2015).
(http://www.sciencedirect.com/science/article/pii/S0021967315014909).
Jack Cochran is a Director of New Business and Technology at Restek Corporation. He is a recognized expert in GC and GC×GC for the analysis of pesticides and priority pollutants. He serves on the Board of Directors for FLAG Works (sponsor of NACRW) and the Centre of Oil and Gas Research and Development at the University of Manitoba. Jack is also an Adjunct Professor in the Forensic Science Program at The Pennsylvania State University.
















University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.

Miniaturized GC–MS Method for BVOC Analysis of Spanish Trees
April 16th 2025University of Valladolid scientists used a miniaturized method for analyzing biogenic volatile organic compounds (BVOCs) emitted by tree species, using headspace solid-phase microextraction coupled with gas chromatography and quadrupole time-of-flight mass spectrometry (HS-SPME-GC–QTOF-MS) has been developed.


