Seeking the Holy Grail—Prediction of Chromatographic Retention Based Only on Chemical Structures
Special Issues


Paul R. Haddad

Over the last decade method development in liquid chromatography (LC) has become an increasingly difficult challenge. Apart from the wide range of chromatographic techniques available, the number of stationary phases that can be used for each technique has increased enormously. Taking reversed-phase LC as an example, there are more than 600 stationary phases from which to choose.
It is convenient to divide method development into two distinct steps. The first is the “scoping” phase, which involves selecting the most appropriate chromatographic technique (reversed-phase LC, hydrophilic-interaction chromatography [HILIC], ion-exchange chromatography, and so on), the stationary phase most likely to yield the desired separation, and the broad mobile-phase composition. The second is the “optimization” phase, wherein the precise mobile-phase composition, flow rate, column temperature, and so forth, are determined. The scoping phase of method development, especially in large industries, has traditionally followed a path whereby a mainstream chromatographic technique is chosen (usually reversed-phase LC), a limited number of stationary phases are then considered (often defined by the range of stationary phases available in the laboratory at the time), and preliminary scouting experiments are performed experimentally on these phases. This approach can prove to be a very costly exercise and often needs to be repeated if, for example, the initial choice of the chromatographic technique was incorrect.
A very attractive alternative would be to conduct the scoping phase of method development by predicting the retention times of the analytes of interest on a range of chromatographic techniques and for a wide range of stationary phases. If this approach were possible, then the selection of chromatographic technique and stationary phase could be made with confidence and all the experimental aspects of method development could be confined to the optimization phase. Note that the accuracy of retention time prediction required for successful scoping need not be excessive, and predictions with errors of about 5% or less could be used for this purpose. In recent years, there has been strong interest in predicting retention of analytes based only on their chemical structures. These studies use quantitative structure-retention relationships (QSRRs), which provide a mathematical relationship between the retention time of an analyte and some properties of that analyte that can be predicted from its structure. These properties may be conventional physicochemical properties, such as molecular weight, molecular volume, polar surface area, logP, logD, and so forth. However, such physicochemical properties are often too limited in number to give accurate prediction models, so researchers frequently use molecular descriptors derived from molecular modeling based on the analyte’s structure. These molecular descriptors may number in the thousands and are derived from one-, two- and three-dimensional (1D, 2D, and 3D) calculations from the chemical structure.
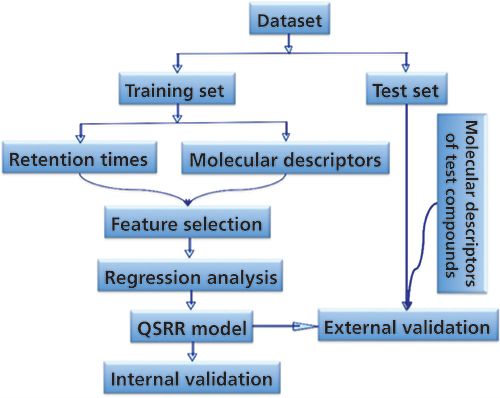
A typical QSRR procedure follows the scheme in Figure 1. First, one needs a database (as large as possible) of known analytes with known structures and retention times for a specified chromatographic condition. As shown in Figure 1, a subset (the training set) consisting of the most relevant analytes in the database is selected and is used to build the desired QSRR mathematical model. One means to select the training set of analytes is to use only those analytes that are chemically similar (as determined by some mathematical similarity index) to the target compound for which retention is to be calculated. Because the number of available molecular descriptors can be very high, it is usually necessary to find those descriptors that are most significant in determining retention. This is called the feature selection step. Finally, the developed model needs to be validated to determine its accuracy of prediction. After the QSRR model has been validated it can then be used to calculate retention time of a new compound simply by calculating the relevant molecular descriptors for that compound (using only its chemical structure) and inserting those descriptors into the model. It should be noted that there is no attempt to derive a “universal” QSRR model applicable to all analytes. Rather, a new “local” model is calculated for each new analyte by identifying the best training set for that specific analyte.

Figure 1: Typical procedure followed in QSRR modeling.
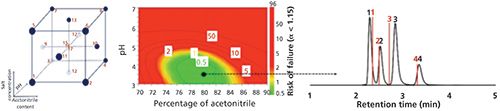
The above process works well, and Figure 2 shows an example wherein the prediction of retention for three groups of four analytes on a HILIC amide stationary phase was performed with excellent accuracy. Retention times for 13 benzoic acids were measured at each experimental point in the experimental design shown on the left. Retention data were predicted for four new benzoic acid analytes at each of the mobile-phase compositions in the experimental design. These predicted retention times were used to construct a color-coded risk of failure map (using a failure criterion of α < 1.15) with the optimal combination of pH and % acetonitrile shown (center), while on the right is the actual chromatogram obtained at the predicted optimal mobile phase, together with the predicted retention times shown as red lines (1). The average error of prediction was 3.8 s, and this level of accuracy was obtained for analytes never seen in the modeling step and at a mobile-phase composition that had not been used in the experimental design.

Figure 2: Output of QSRR modeling for HILIC separations on a Thermo Fisher Acclaim HILIC-10 amide column. See text for discussion. The experimental chromatogram (black peaks) corresponds to the selected optimal mobile-phase composition and the red lines represent the predicted retention times from the QSRR-design of experiment (DoE) models. Numbering of test compounds: 1 = 4-aminobenzoic acid, 2 = 3-hydroxybenzoic acid, 3 = 4-aminosalicylic acid, 4 = 2,5-dihydroxybenzoic acid. Adapted with permission from reference 3.
It is clear that QSRR modeling can achieve the level of accuracy desired for the scoping phase of method development. However, there are some important limitations. The first of these is the need for extensive retention data. To be successful, the QSRR modeling requires that the database contains sufficient chemically similar compounds to enable identification of a training set of at least 7−10 compounds. Second, if the database contains retention data measures for one stationary phase at one mobile-phase composition then all predictions of retention can be made only under those conditions. The second limitation can be overcome by changing the target of the model from retention time to some fundamental parameters that allow calculation of retention time. One example of this approach is in ion-exchange chromatography, where retention is governed by the linear solvent strength model (2):
log k = a - b log [Ey-] [1]
where k is the retention factor, [Ey-] is the molar concentration of the eluent competing ion (mol/L), and a and b values are the intercept and the slope, respectively. If QSRR models are built for the a- and b-values in equation 1, then retention factor (and retention time) can be calculated for any mobile-phase composition. The same approach can be used for other chromatographic techniques, such as by modeling the analyte coefficients for the hydrophobic subtraction model in reversed-phase LC (3).
The first limitation discussed above remains the biggest obstacle to successful implementation of QSRR modeling, especially in reversed-phase LC because of the huge diversity of analytes and stationary phases involved. The creation of sufficiently diverse and reliable databases will be beyond the reach of any individual organization and will be possible only through extensive networking and collaborations. Crowd sourcing data might be one way to acquire the data, but the requirements for structured procedures and validated results will probably render this approach impractical. On the other hand, consortium arrangements between large industrial organizations, such as pharmaceutical companies, could be the solution since these organizations share the same general method development problems and they are accustomed to the stringent requirements for the accuracy of chromatographic data.
Until sufficient databases are generated, the full potential of QSRR modeling will not be realized and the “Holy Grail” will remain elusive. However, the opportunity to predict retention solely from chemical structure remains a highly desirable outcome.
Acknowledgment
The author acknowledges the contributions to this project from the following collaborators: Maryam Taraji (UTAS), Soo Park (UTAS), Yabin Wen (UTAS), Mohammad Talebi (UTAS), Ruth Amos (UTAS), Eva Tyteca (UTAS), Roman Szucs (Pfizer), John Dolan (LC Resources), and Chris Pohl (Thermo Fisher Scientific).
References
- M. Taraji, P.R. Haddad, R.I.J. Amos, M. Talebi, R. Szucs, J.W. Dolan, and C.A. Pohl, Anal. Chem. 89, 1870–1878 (2017).
- S. Park, P.R. Haddad, M. Talebi, E. Tyteca, R.I.J. Amos, R. Szucs, J.W. Dolan, and C.A. Pohl, J. Chromatogr. A 1486, 68–75 (2017).
- L.R. Snyder, J.W. Dolan, and P.W. Carr, J. Chromatogr. A 1060, 77 (2004).
Paul R. Haddad is an emeritus distinguished professor with the Australian Centre for Research on Separation Science (ACROSS) at the University of Tasmania in Tasmania, Australia.


























Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

