Retention and Selectivity of Stationary Phases Used in HILIC
LCGC Europe
The retention and selectivity of stationary phases in HILIC is discussed.
Pages 102-109
Hydrophilic-interaction chromatography (HILIC) is a complex system involving partition, polar, and ion-exchange interactions. Method development can be greatly facilitated by understanding the interactions that the different stationary phases provide and applying that knowledge to the separation task at hand. Through an understanding of the main differences of the set of analytes to be separated, a column or set of columns can be judiciously chosen to screen for effective retention and selectivity.
Analogous to reversed-phase chromatography, polar stationary phases used in hydrophilic-interaction chromatography (or HILIC as coined by Alpert [1]) provide different interactions that can be exploited by chromatographers to retain and separate various components of a mixture. Mechanisms of interaction in HILIC include partitioning, polar interactions, and ionic interactions (2). Partitioning involves the phase transfer of polar analytes from an organic-rich mobile phase into an adsorbed layer of water on the stationary phase. Polar interactions may then occur between the active surface or ligands of the stationary phase and ionic interactions may occur between charged analytes and oppositely charged moieties on the stationary phase. Stationary-phase chemistries can be designed to heighten or attenuate the various mechanisms and thus impart alternative retention and selectivity. For example, bare silica has been shown to adsorb water in the presence of an organic-rich mobile phase and thus provides the opportunity for an analyte to partition. Bare silica also exhibits negatively charged silanols at moderate pH conditions and above that may interact strongly with positively charged analytes (ion-exchange) (3). A penta-fluorophenyl phase has been shown to exhibit very little partition, yet provides a high degree of ion-exchange potential (4). On the other end of the spectrum, a pentahydroxyl stationary phase predominantly retains analytes though partition mechanisms because of the high degree of water adsorbed by the surface and shows relatively little ion-exchange capacity. Through an understanding of the interactions stationary-phase chemistries provide, one can choose the most appropriate blend of interactions that best complements a given separation challenge.
HILIC is characterized by the use of a polar stationary phase and a relatively nonpolar mobile phase containing a small amount of water (1). The more polar water preferentially absorbs on the polar stationary phase providing an environment conducive to partitioning mechanisms. If a polar molecule, then, finds itself residing in the relatively nonpolar mobile phase it will tend to partition into the more desirable polar aqueous realm. Conditions are thus adjusted by chromatographers to induce both partitioning into and out of the aqueous layer. The system is pictorially shown in Figure 1.


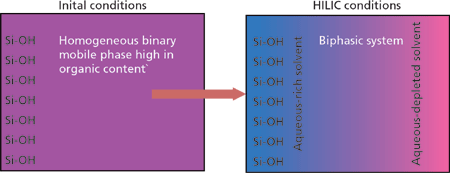
Figure 1: Biphasic solvent distribution at the HILIC phase surface.
Unfortunately, the situation is not that simple. By definition, polar molecules are interactive. Polar molecules are often ionic and thus may interact via ion-exchange mechanisms in addition to partitioning. At moderate pH and higher, silica-based phases exhibit negatively charged ionized silanols that may interact strongly with positively charged analytes. Negatively charged analytes may also be repulsed. Polar molecules may exhibit strong dipole moments or hydrogen bonding moieties that interact via adsorptive polar interactions. In HILIC, these highly interactive molecules are drawn very close to another polar, highly interactive surface making ion-exchange and polar interactions highly probable.
Polar and ionic interactions are mechanisms that exist in the more familiar reversed-phase chromatography; however, these interactions are generally regarded as secondary and thus less important to partitioning. In HILIC, however, polar and ionic interactions are significant contributors to overall retention and selectivity and, in fact, are often the main mechanisms of interaction.


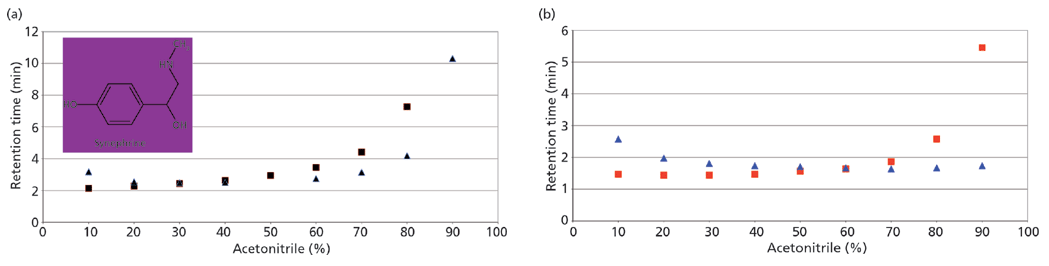
Figure 2: Retention profile of synephrine on bare silica (squares) and pentafluoro-phenyl (triangles) phases: (a) buffer diluted as organic is added; (b) buffer held constant. Mobile phase: 13 mM ammonium acetate (pH 6.7)–acetonitrile.
Multiple Interactions in HILIC
Figure 2(a) shows a retention profile for the polar basic compound synephrine using two different HILIC stationary phases. In this figure, the retention is shown as a function of increased acetonitrile in the mobile phase and consequently the dilution of the buffer concentration. On both phases, the retention of synephrine is shown to increase at high acetonitrile content and thus both systems appear to be "HILIC". The plot in Figure 2(b) shows the same experiment, but the buffer concentration of the mobile phase was kept constant as the organic modifier was increased. This approach has a dramatic effect on one column and virtually no effect on the other. What this demonstrates is that all retention under HILIC conditions is not necessarily because of partitioning mechanisms. On the bare silica phase, water is indeed adsorbing on the surface and thus providing the opportunity for the polar synephrine to partition. The retention is not impacted to a large extent by the change in buffer concentrations. On the pentafluorophenyl phase, however, there appears to be little or no water on the surface. Retention is therefore based on polar or ionic interactions and is highly influenced by the buffer concentration.
The variables used to control and adjust partition and ionic interactions are very different. It is therefore paramount to have some understanding of the interactions that take place in a given system. It is also important to have a firm grasp on the interactions different stationary-phase chemistries provide to make sound choices during method development. As in reversed-phase chromatography, understanding retention mechanisms in HILIC can greatly facilitate method development and result in robust and rugged methods (5).
For any analysis where the potential for ion-exchange exists, a good general practice is to evaluate the ion-exchange contribution to the overall retention and selectivity. For an ion-exchange process involving a singly charged analyte, the dependence of retention on the mobile-phase competing ion may be expressed as follows (6):


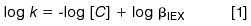
where k is the capacity factor of the analyte, C is the concentration of the competing mobile-phase modifier, and βIEX is a constant for a given system that includes the phase ratio, ion-exchange capacity of the stationary phase, and the ion-exchange equilibrium constant. A plot of log k versus log[C] will thus yield a slope of -1 when ion exchange is solely responsible for the retention of the analyte, whereas the plot would yield a slope of 0 where ion exchange is not present.


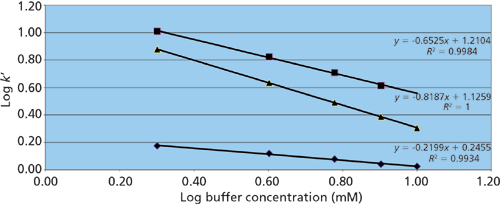
Figure 3: Response of ephedrine retention on buffer concentration on three HILIC phases. Phases: Pentafluorophenyl, squares; bare silica, triangles; pentahydroxyl, diamonds. Mobile phases for all columns were 2, 4, 6, 8, and 10 mM ammonium acetate (pH unadjusted) in 90% acetonitrile.
Figure 3 shows the results of such a study on Ascentis Express F5 (pentafluorophenyl), Ascentis Express OH5 (pentahydroxyl), and Ascentis Express HILIC (bare silica) stationary phases (all from Supelco) using ephedrine, a polar basic analyte, as the solute. On the pentafluorophenyl phase the resulting slope approaches a value of -1 indicating a dominant contribution to retention from ion-exchange. The pentahydroxyl phase, however, exhibits a slope nearing 0 indicating relatively little ion-exchange character. The bare silica phase shows a slope in the mid-range indicating a significant contribution from ion exchange, but also a significant contribution from other mechanisms. Because polar interactions are difficult to isolate, the "rest" of the retention can be deemed to be caused by a combination of polar and partitioning mechanisms. Table 1 provides a list of some common HILIC phases and the resulting ion-exchange contributions from a similar study using normetanephrine as a probe. In addition, the table shows the retention of cytosine, a neutral analyte under the run conditions that is indicative of the relative partition and polar interaction contributions for each phase.


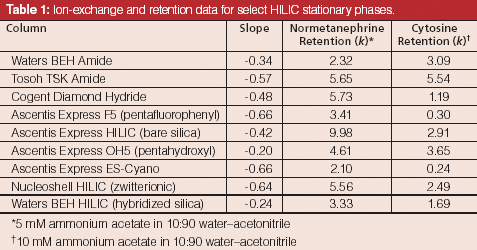
Table 1: Ion-exchange and retention data for select HILIC stationary phases.
The pentafluorophenyl and cyano phases both show relatively high ion-exchange contributions to the normetanephrine retention along with little partitioning or polar interactions as noted by the lack of cytosine retention. This finding is consistent with recent studies of Soukup and Jandera (7), where they used coulometric Karl-Fischer measurements to determine preferential uptake of water for different HILIC stationary phases. Simply stated, the results show that phases like cyano and pentafluorophenyl exhibit ion exchange, but relatively little partitioning.
On the other end of the spectrum lie the pentahydroxyl and BEH Amide (Waters) phases. Both stationary phases exhibit low ion-exchange and relatively high partition or polar interactions as demonstrated by the near zero slope values and the strong retention for cytosine, respectively. These findings are consistent with a study published by McCalley (3) under relatively high buffer concentrations.
The bare silica phase showed moderate ion-exchange contributions along with moderate retention for cytosine. This is again consistent with the other works cited. The hybridized silica demonstrated low ion-exchange contributions, presumably because of the lower number of silanol groups present. The hybridized silica column did exhibit good retention for cytosine and thus represents a different relative contribution of ion-exchange and partition or polar interactions.
Surprisingly, the Diamond Hydride phase (Cogent) showed moderately strong ion-exchange character. The Diamond Hydride phase is built on type-C silica that is largely populated with silicon-hydride (Si-H) groups instead of silanol groups (Si-OH) as found on traditional silica surfaces (8). With the majority of the surface rendered nonionic, low ion-exchange behaviour would be expected. The relatively strong ionic character observed remains unexplained, but is consistent with the findings of Bawazeer and colleagues (9). The Diamond Hydride phase exhibits moderate retention for cytosine and thus represents another useful blend of the various retention mechanisms.
Also surprising were the relatively high ion-exchange contributions from both the Tosoh Amide (Tosoh Bioscience) and the Nucleoshell HILIC (zwitterionic) (Macherey Nagel) phases. Amide phases are generally characterized by low ion-exchange and high partition or polar interactions; however, based on the present study the Tosoh Amide phase appears to exhibit relatively high ion-exchange and high partitioning or polar contributions. Similarly, zwitterionic phases tend to have low ionic interactions and high partition or polar interactions, while the Nucleoshell column shows high ionic character and only moderate partitioning or polar mechanisms. These findings may be because of the conditions under which they are obtained, the manner in which the phases are bonded, or the chemical properties of the surfaces they are built on. Needless to say, one cannot conclude that all "amide", "zwitterionic", or especially "HILIC" phases provide the same blend of interactions.
Considerations for HILIC Method Development
There are many different sets of analytes one may wish to apply to HILIC chromatography. A good starting point for method development is to identify the stationary phase with the appropriate mechanisms that best retain and differentiate the analytes. At a minimum, understanding the attributes of the stationary phase can be used to eliminate those phases that are inappropriate. For example, if a set of polar neutral molecules are to be separated, polar and partitioning mechanisms will be required whereas ionic interactions would be ineffective. Using a pentafluorophenyl stationary phase that primarily exhibits ion exchange would then be a wasted effort. The key to using any chromatographic mode is to have some idea of where the greatest differences lie with respect to the different analyte physiochemical attributes and invoke the most selective retention mechanisms by choosing the most appropriate phase or phases to screen.
For method development in HILIC, a sound approach is to
- Understand the analyte properties. (Will they partition? Are they polar enough to partition into the aqueous phase? Will they ion-exchange? Are they charged, positively or negatively? How do they differ most, solubility (log P/log D) or degree of ionization (pKa values)?
- Choose the most appropriate stationary phase or phases that complement the largest differences in the analytes or, conversely, eliminate those that do not.
- Screen the chosen phases for the best selectivity.
- Optimize the separation using the most appropriate column chemistry.


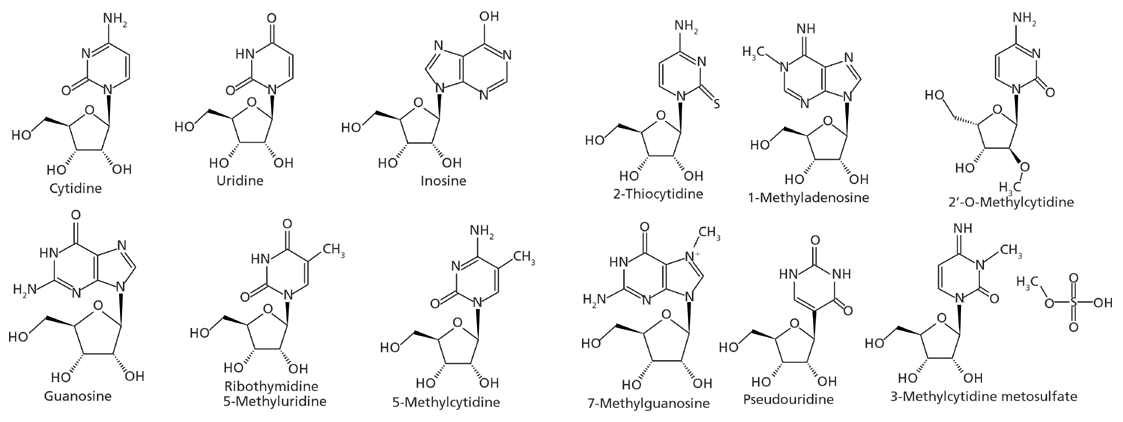
Figure 4: Nucleoside structures (polar neutral for the most part).
The development of a HILIC separation for a set of nucleosides is used to demonstrate the utility of this approach (10). Nucleosides in general are polar molecules and are weakly basic, thus falling under the category of "polar neutrals." Cytidine, for example, exhibits a basic pKa value of 4.3 whereas its most acidic pKa is 13.5. Below a pH of 4.3 the compound would predominantly carry a positive charge, but under most HILIC conditions, the effective pH is such that the compound will be neutral. Figure 4 and Table 2 present the structures of the study analytes and pertinent physicochemical data, respectively. The set of compounds were screened using bare silica, pentahydroxyl, and pentafluorophenyl phases under several sets of HILIC conditions. As one would predict, only those phases exhibiting partition mechanisms proved useful.


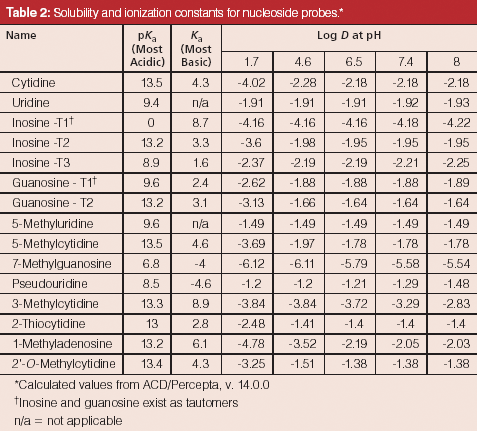
Table 2: Solubility and ionization constants for nucleoside probes.*
The majority of the nucleosides are neutral within the useful chromatographic pH window and thus cannot interact via ion exchange. To provide retention for the polar neutral molecules, the stationary phase must provide a partitioning mechanism. Both the pentahydroxyl and bare silica phases are polar enough to adsorb water onto their surfaces, thus enabling the potential for partitioning to take place. Figures 5 and 6 show the screening results for the pentahydroxyl and bare silica columns, respectively. Both phases provide good retention and selectivity for all of the probes. It is interesting to note that the last three compounds eluted show increased relative retention on the bare silica phase as compared to the pentahydroxyl phase. The retention of these late-eluted compounds also varies with pH on the bare silica phase, but their retention is relatively stable using the pentahydroxyl system. Both observations indicate significant ion exchange is occurring on the bare silica phase and demonstrates the limited ion exchange exhibited by the pentahydroxyl phase. The late eluted compounds were identified as 1-methyladenosine (basic pKa 6.1), 3-methycytidine (basic pKa 8.9), and the permanently positively charged 7-methylguanosine. Figure 7 shows the same experiments run on the pentafluorophenyl phase for demonstration purposes. As expected, little or no retention is observed for the polar neutral molecules because of the lack of partitioning provided by this particular phase chemistry. Again, it appears that a few of the nucleosides are charged enough to retain on the pentafluorophenyl phase via ion-exchange mechanisms.


Figure 5: Screening nucleosides on a pentahydroxyl phase.
The screening data indicated that the pentahydroxyl column using a pH of 5 provided the most selective and efficient starting point for further method development. These conditions were slightly refined to provide the useful separation shown in Figure 8. As per Table 1, column chemistries such as the Waters BEH HILIC and BEH Amide that show low ion-exchange interactions and significant partitioning might also be expected to produce similar retention and selectivity. This has indeed been found for the BEH Amide phase by Li and colleagues (11).


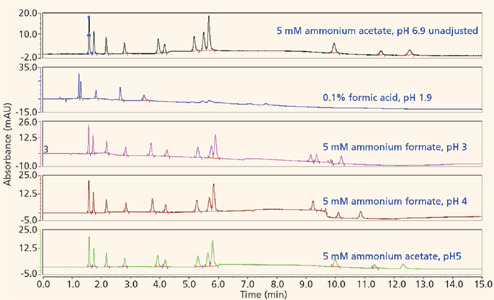
Figure 6: Screening nucleosides on a bare silica phase.
Many examples of this approach can be found in the literature. For example ephedrine and related compounds were separated under liquid chromatography–mass spectrometry (LC–MS)-compatible conditions via primarily ion-exchange mechanisms on a pentafluorophenyl phase (12). For the separation of cathinone-derived designer drugs, both partition and polar interactions along with ionic mechanisms were required to resolve isobaric analytes (13).


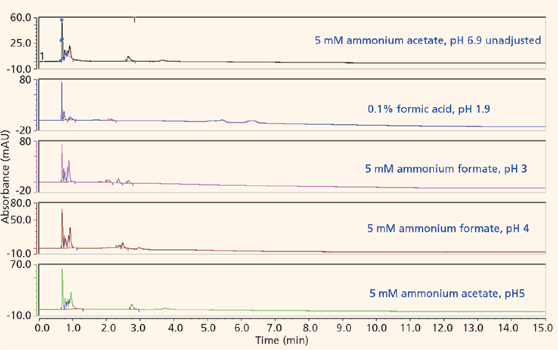
Figure 7: Screening nucleosides on a pentafluorophenyl phase.
Conclusions
HILIC chromatography is a complex system involving partition, polar, and ion-exchange interactions. Method development can be greatly facilitated by understanding the interactions that the different stationary phases provide and applying that knowledge to the separation task at hand. A straightforward approach towards determining the relative ion-exchange contribution for various HILIC stationary phases has been described and can be used to classify HILIC columns by their predominant mechanisms of interaction.


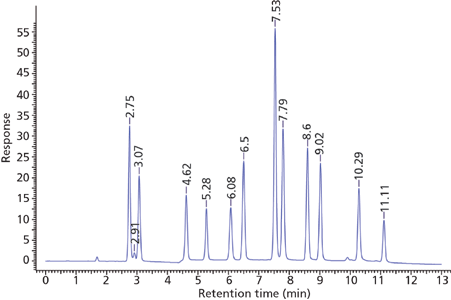
Figure 8: Optimized separation of nucleosides. Column: 10 cm × 2.1 mm, 2.7 μm dp Ascentis Express OH5 (Sigma-Aldrich); mobile-phase A: 5 mM ammonium acetate, adjusted to pH 5.0 with acetic acid, in 95:5 acetonitrile–water; mobile phase B: 5 mM ammonium acetate, adjusted to pH 5.0 with acetic acid, in 80:20 acetonitrile–water; gradient: 0% B held for 1 min, 0–100% B in 10 min, held at 100% B for 1 min; flow rate: 0.3 mL/min; column temperature: 25 °C; detection: UV absorbance at 250 nm; injection volume: 2 μL; sample concentration: 10–100 μg/mL. Peaks: ribothymidine, 2.75 min; uridine, 3.07 min; 2-thiocytidine, 4.62 min; 2'-O-methylcytidine, 5.28 min; pseudouridine, 6.08 min; inosine, 6.50 min; 5-methylcytidine, 7.53 min; cytidine, 7.79 min; guanosine, 8.60 min; 3-methylcytidine, 9.02 min; 1-methyladenosine, 10.29 min; 7-methylguanosine, 11.11 min.
The investigation demonstrated that diol/pentahydroxy and BEH Amide phases provide relatively little ion-exchange interactions and relatively high partitioning and polar interactions. Conversely, pentafluorophenyl and cyano phases exhibit primarily ion-exchange and relatively little partitioning character. Bare silica and several variations of HILIC columns are shown to provide different blends of ion-exchange, partitioning, and polar interactions. For a neutral set of molecules, such as the nucleosides, only partition can be expected to provide interactions that result in retention and selectivity. Indeed, the pentahydroxyl and the bare silica phase were shown to be useful and worth the time to investigate. Through an understanding of the main differences of the set of analytes to be separated, a column or set of columns can be judiciously chosen to screen for effective retention and selectivity.
David S. Bell is a manager in pharmaceutical and bioanalytical research at Sigma-Aldrich/Supelco. With a B.S. degree from SUNY Plattsburgh and a PhD in Analytical Chemistry from The Pennsylvania State University, Dave spent the first decade of his career within the pharmaceutical industry performing analytical method development using various forms of chromatography and electrophoresis. During the past 15 years, working directly in the chromatography industry, Dave has focused his efforts on the design, development, and application of stationary phases for use in HPLC and hyphenated techniques. In his current role at Supelco, Dave's main focus has been to research, publish, and present on the topic of molecular interactions that contribute to retention and selectivity in an array of chromatographic processes. Direct correspondence to: dave.bell@sial.com
Ronald E. Majors is the editor of "Column Watch," an analytical consultant, and a member of LCGC Europe's editorial advisory board. Direct correspondence about this column should be addressed to "Column Watch", LCGC Europe, Honeycomb West, Chester Business Park, Wrexham Road, Chester, CH4 9QH, UK, or e-mail the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com
References
(1) A.J. Alpert, J. Chromatogr.499, 177–196 (1990).
(2) D.V. McCalley in Hydrophilic Interaction Chromatography: A Guide for Practitioners, B.A. Olsen and B.W. Pack, Eds., (John Wiley & Sons, Hoboken, New Jersey, USA, 2013), pp. 1–41.
(3) D.V. McCalley, J. Chromatogr. A1217, 3408–3417 (2010).
(4) D.S. Bell and A.D. Jones, J. Chromatogr. A 1073, 99–109 (2005).
(5) C.R. Aurand, H. Cramer, J. McKenzie, and D.S. Bell, LCGC North Am.27(9), 493–502 (2014).
(6) X. Yang, J. Dai, and P.W. Carr, J. Chromatogr. A 996, 13–31 (2003).
(7) J. Soukup and P. Jandera, J. Chromatogr. A1374, 102–111 (2014).
(8) J.J. Pesek, M.T. Matyska, R.I. Boysen, Y. Yang, and M.T.W. Hearn, TrAC Trends in Anal. Chem.42, 64–73 (2013).
(9) S. Bawazeer, O.B. Sutcliffe, M.R. Euerby, S. Bawazeer, and D.G. Watson, J. Chromatogr. A1263, 61–67 (2012).
(10) D.S. Bell and H. Cramer, Supelco Reporter32.1, 6–8 (2014).
(11) F. Li, J.A. Duan, D. Qian, S. Guo, Y. Ding, X. Liu, Y. Qian, Y. Peng, Y. Ren, and Y. Chen, J. Pharm. Biomed. Anal. 83, 10–18 (2013).
(12) D.S. Bell, H.M. Cramer, and A.D. Jones, J. Chromatogr. A 1095, 113–118 (2005).
(13) X. Li, C.E. Uboh, L.R. Soma, Y. Liu, F. Guan, C.R. Aurand, D.S. Bell, Y. You, J. Chen, and G.A. Maylin, Rapid Commun. Mass Spectrom. 28, 217–229 (2014).













Assessing Thorium-Peptide Interactions Using Hydrophilic Interaction Liquid Chromatography
February 4th 2025Paris-Saclay University scientists used hydrophilic interaction liquid chromatography (HILIC) coupled to electrospray ionization mass spectrometry (ESI-MS) and inductively coupled plasma mass spectrometry (ICP-MS) to assess thorium’s interaction with peptides.



