Rapid Sample Preparation Followed by a 100% Water Mobile Phase HPLC Analysis for Quantifying Acetamiprid and its N-desmethyl Metabolite, IM-2-1, in Cow’s Milk
This article describes a space-saving, quick, and inexpensive sample preparation technique followed by a high performance liquid chromatography (HPLC) method with a 100% water mobile phase and photodiode array (PDA) detection for quantifying acetamiprid and its N-desmethyl metabolite, IM-2-1, in cow’s milk. The analytes were extracted from the sample and deproteinized using a handheld ultrasonic homogenizer with 5% (w/v) trichloroacetic acid solution, purified using a centrifugal monolithic silica spin minicolumn, and quantified within 20 min per sample. The accuracy and precision are well within the international method acceptance criteria.


Photo Credit: Iliveinoctober/Shutterstock.com

Naoto Furusawa, Graduate School of Human Life Science, Osaka City University, Osaka, Japan


This article describes a space-saving, quick, and inexpensive sample preparation technique followed by a high performance liquid chromatography (HPLC) method with a 100% water mobile phase and photodiode array (PDA) detection for quantifying acetamiprid and its N-desmethyl metabolite, IM-2-1, in cow’s milk. The analytes were extracted from the sample and deproteinized using a handheld ultrasonic homogenizer with 5% (w/v) trichloroacetic acid solution, purified using a centrifugal monolithic silica spin minicolumn, and quantified within 20 min per sample. The accuracy and precision are well within the international method acceptance criteria.
In the early 2000s several kinds of neonicotinoids, a class of neuroactive–systemic insecticides, began to come under increasing scrutiny because of their potential environmental impact. The use of neonicotinoids was linked in a range of studies to a number of adverse ecological effects, including honeybee colony collapse disorder and loss of birds because of a reduction in insect populations. Increased scrutiny eventually led to restrictions and bans on the use of different neonicotinoids in several countries (1–3).
In December 2013, the European Food Safety Authority (EFSA) disclosed that two neonicotinoid insecticides, including imidacloprid and acetamiprid, a widely and frequently used neonicotinoid insecticide, may affect the developing human nervous system. Experts from the Authority propose that guidance levels for acceptable exposure to the two neonicotinoids be lowered while further research is performed to provide more reliable data on so-called developmental neurotoxicity (4).
Previous acetamiprid metabolism studies on animals (rats, lactating goats, and laying hens) have found that the main metabolic pathway of acetamiprid in rats is the transformation to N-desmethyl acetamiprid, IM-2-1 (IM2); the predominant residue in most animal products (goat milk, goat liver or kidney, and hen’s liver, muscle, and skin) is the IM2 (5). To ensure the safety of animalâderived foods for the consumer, the Japanese Health, Labour, and Welfare Ministry (JHLWM) sets maximum residue limits (MRLs) for the sum of acetamiprid and IM2, expressed as acetamiprid (6). The determination of acetamiprid and IM2 in animalâderived foods is therefore important to guarantee food safety, and a validated analytical method for the simultaneous determination of acetamiprid and IM2 is currently required.
Depending on the recent expansion and diversification in the international food trade, the development of international harmonized methods to determine chemical residues in foods is essential to guarantee equitable international trade in these foods and ensure food safety for consumers. An international harmonized analytical method for residue monitoring in foods is urgently needed. The optimal harmonized method must be easy to use and economical in time and cost, and must cause no harm to the environment and analyst.
Although several methods have been described in the literature to determine acetamiprid and IM2 in honey, mice, or human serum and urine (7–12), these methods have three crucial drawbacks: first, they involve several analytical steps in the sample preparation, which are time- and cost-consuming and do not permit the determination of large number of samples; and second, all of the methods consume large quantities of organic solvents, acetonitrile or methanol, in the mobile phases. Risk associated with these solvents extend beyond direct implications for the health of humans and wildlife to affect the environment and the ecosystem. Eliminating the use of organic solvents is an important goal in terms of environmental conservation, human health, and the economy (13). The third drawback is that most of the recent methods are based on liquid chromatography–mass spectrometry (LC–MS) or LC–MS/MS. LC–MS/MS systems are mainly available in industrial nations because they are hugely expensive, and the methodologies are complex and specific. These systems are unavailable in a lot of laboratories for routine analysis, particularly in developing countries. No optimal method that satisfies the aforementioned requirements has yet been identified.
This article describes a technique that can be adopted as an international harmonized analytical method for the residue monitoring of acetamiprid and IM2 in animalâderived foods. The technique comprises a simplified space-saving sample preparation with reduced organic solvent consumption followed by HPLC with an isocratic 100% water mobile phase and photodiode-array (PDA) detection for determining acetamiprid and IM2 in cow’s milk. Milk contains a good balance of protein, fat, and carbohydrate, and it is an indispensable food because it is inexpensive and readily available. The JHLWM has set the MRL for acetamiprid (including IM2) in milk at 0.1 ppm (6).
Experimental
Reagents and Apparatus: All chemicals including acetamiprid and IM-2-1 (IM2, N-desmethyl acetamiprid) standards were purchased from Wako Pure Chem. Ltd. 100% (w/v) trichloroacetic acid (TCA) was used as a deproteinization agent. Ethanol (non-toxic class, the human or environmental toxicity is negligible [14]) and distilled water were of HPLC grade.
The following apparatus was used in the sample preparation: a handheld ultrasonic-homogenizer (model HOM-100, 2-mm i.d. probe, Iwaki Glass Co., Ltd.); a micro-centrifuge (Biofuge fresco, Kendo Lab. Products); three types of centrifugal monolithic silica spin minicolumns (13 mm × 26.8 mm) (sample throughput volume ≤800 μL), including a MonoSpin C18 column (bonded with an octadecyl group), a MonoSpin C18âAX column (bonded with an octadecyl group and a quaternary ammonium group), and a MonoSpin C18-CX column (bonded with an octadecyl group and a benzene sulfonic acid group) (all GL Sciences, Inc.).
The HPLC system used for method development included a model PU-980 pump and DG-980â50âdegasser (Jasco Corp.) equipped with a model CTO-10AS VP column oven (Shimadzu Scientific Instruments), as well as a model SPD-M10A VP photodiode-array detector (Shimadzu).
HPLC Operating Conditions: The analytical column was a 150 × 4.6 mm, 5-μm Inertsil WP300 C4 column (GL Sciences) using an isocratic mobile phase of water at a flow rate of 1.0 mL/min at 50 °C. The PDA detector was operated at 190–350 nm: the monitoring wavelengths were adjusted to 234 nm and 245 nm, which re present maximums for acetamiprid (at 245 nm) and IM2 (at 234 nm), respectively. The injection volumes were 10–20 μL.
Preparation of Stock Standards and Working Mixed Solutions: Stock standard solutions of acetamiprid and IM2 were prepared by dissolving each compound in water followed by water to a concentration of 50 μg/mL. Each solution was stored at -20 °C. Working mixed standard solutions of these two compounds were freshly prepared by suitably diluting the stock solutions with water on the day of the analysis.
Preparation of Calibration Standards and Quality Control Samples: For method validation studies, calibration standards and quality control samples (QCs), which are terms defined in the US Food and Drug Administration (FDA) guideline (15), were prepared by spiking appropriate aliquots of the mixed standard solution in blank milk samples. Calibration standards were used to construct calibration curves from which the concentrations of analytes in unknown monitoring samples could be determined. QCs were used to evaluate the performance of the proposed method. In this study, the standards were prepared in the range of 0.1 μg/mL, 0.2 μg/mL, 0.5 μg/mL, 1.0 μg/mL, 2.0 μg/mL, and 3.0 μg/mL for both analytes. Three QC levels (for both analytes, QC1 = 0.1 μg/mL; QC2 = 0.5 μg/mL; QC3 = 2 μg/mL) were prepared.
Sample Preparation: An accurately measured 100-μL milk sample was taken into a 1.5-mL micro-centrifuge tube and homogenized with 600 μL of 5% (w/v) TCA solution with a handheld ultrasonic-homogenizer for 30 s. After being homogenized, the capped tube was centrifuged at 14,180 g for 5 min. A 100 μL of supernatant liquid was poured into a MonoSpin C18-CX minicolumn, and the capped minicolumn was centrifuged at 1030 g for 1 min. Using a similar centrifuging operation, the minicolumn was then washed with 100 μL of 5% (v/v) ethanol (in water) and then acetamiprid and IM2 was eluted with 100 μL of 25% ethanol. The eluate was injected into the HPLC system.
Method Validation: The performance of the developed method was validated in terms of many parameters from the international guidelines for bio-analytical procedures (16–19).
Results and Discussion
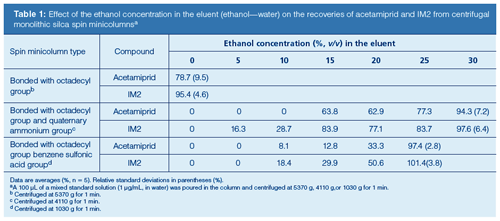
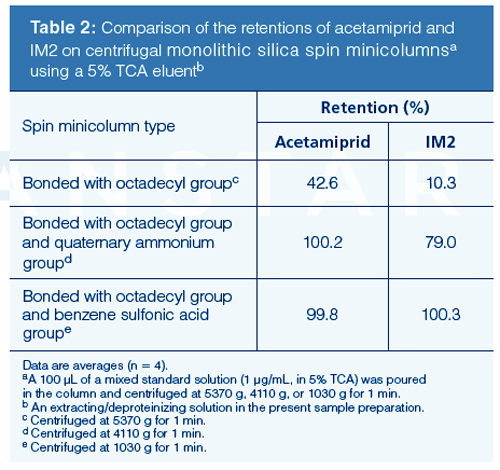
Sample Preparation: The procedure described in this article is a very easy and small-scale technique that reduces organic solvent consumption in the preparation of analytes. The ultrasonic-homogenization enabled the satisfactory extraction of acetamiprid and IM2 from a milk sample with 5% TCA solution. The extract obtained by this operation was purified using a centrifugal monolithic silica spin minicolumn. The spin minicolumn is a monolithic solid-phase extraction (SPE) column designed for use with small sample volumes and a centrifuge (20). The author used three types of the spin minicolumns, and the retention profiles of analytes in these columns were compared.
Since the collection of the eluent from respective spin minicolumn varies with centrifugal acceleration (rotary speed), the author investigated in advance the optimum acceleration (>84 g) to collect the full content of the eluent from each minicolumn. In this test, water was used as an eluent and centrifuged as described earlier. A 100-μL measure of water was applied to the spin minicolumn. The centrifugal time was standardized at 1 min. The optimal accelerations that collected full content of water from the three spin mini-columns were 5370 g, 4110 g, or 1030 g (see footnote in Table 2).
The possibility of eluting analytes from the columns using various eluents was evaluated. Since the analytes were eluted from the minicolumns using polar solvents (normally toxic solvents like acetonitrile and methanol are used) added to the eluent, the effect of the concentration of ethanol (= non-toxic class [14]) in the eluent (ethanol/water, v/v) on the recovery of acetamiprid and IM2 standards from the minicolumns was determined when the centrifugal accelerations were performed at the optimal accelerations for these minicolumns, respectively. The time was standardized at 1 min. In this study, a 100-μL measure of a mixed standard solution containing 0.1 μg of each compound was applied to the spin minicolumn and the eluate was determined by HPLC. The resulting profiles are given in Table 1. As shown, the column with the sorbent modified with an octadecyl group and benzenesulfonic acid showed the best recoveries (≥97.4%) and repeatabilities (relative standard deviations ≤3.8%) for acetamiprid and IM2 when the eluent was 25% ethanol. Based on the findings in Table 2, that column was used as a centrifugal monolithic silica spin minicolumn. Because both analytes were not eluted with 5% ethanol from the spin minicolumn, 5% ethanol was used as the washing eluent to eliminate any interfering compounds of sample origin (Table 1). This isolated protein and fat constituents from the column. The present procedure yields a smallâscale extraction and easy purification of acetamiprid and IM2 in a short time, while significantly limiting the consumption of organic solvents (TCA + ethanol 60 μL/sample). The time required for the sample preparation of a single sample was <10 min. The procedure resulted in high recovery and reproducibility.


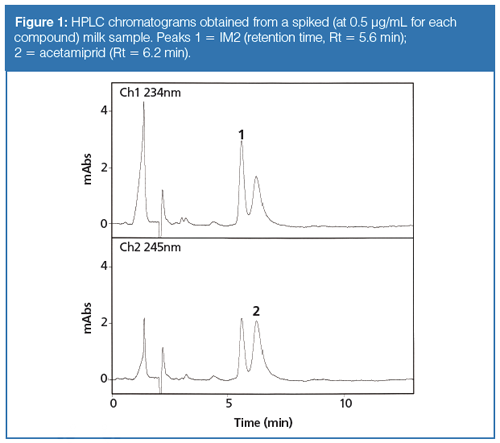
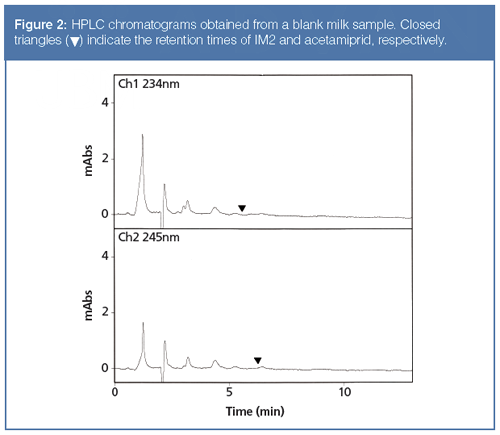
Figures 1 and 2 illustrate that the resulting HPLC chromatograms were free of interfering compounds for the quantification and identification of acetamiprid and IM2; the PDA detector was set at 245 nm for acetamiprid and 234 nm for IM2 (to obtain the maximum absorbance for the two analytes). The HPLC method accomplished good separation with the need for a gradient system to improve the separation. These figures demonstrate that the present method can provide the quantification and identification of the analytes.


Method Validation:
Main Method Validation Data: Table 3 summarizes the main method validation parameters. The quantitative limits in milk samples were 0.049 μg/mL for acetamiprid and 0.033 μg/mL for IM2, respectively. The systemâsuitability evaluation is an essential parameter of HPLC determination, and it ascertains the strictness of the system used. The suitability was evaluated as the relative standard deviations of peak area and retention time calculated for 10 replicate injections of a spiked milk sample with acetamiprid and IM2 (0.5 μg/mL each). The values for acetamiprid and IM2 were estimated to be <0.01% for retention time and ≤0.04% for peak area, respectively. The accuracy and precision are within the international method acceptance criteria (Table 3) (16–19). The other validation findings are as follows:

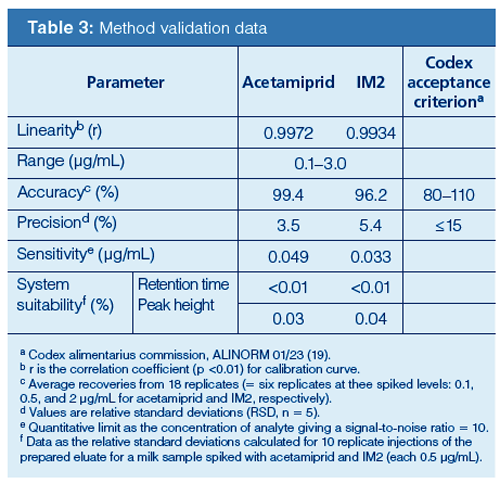
Specificity and Selectivity: The application of the proposed procedure to 10 blank milk samples demonstrated that no interference peak was present near the retention times for acetamiprid and IM2 in any of the samples examined.
The HPLC–PDA method easily confirmed the peak identity of the target compound. Both analytes were identified in a milk sample by their retention times and absorption spectra. The acetamiprid and IM2 spectra obtained from the milk sample were practically identical to those of the standards. Because complete separation of the analytes was achieved, trace-level PDA detection is possible. It is therefore instructive to demonstrate the purification effectiveness of the sample preparation. The system did not require the use of MS or MS/MS, which is very expensive and is not widely available for routine analysis.
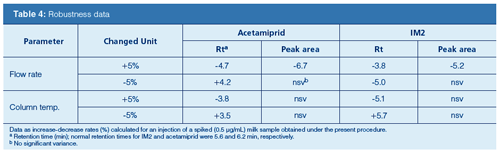
Robustness: In this test, several HPLC parameters were performed using a spiked (0.5 μg/mL of each compound) milk sample obtained under the established procedure. Changes of ±5% units of the flow rate (1.0 mL/min) and the column temperature (50 °C) were determined. The effect on the peak areas and the validations in the retention times were evaluated. Normal retention times for acetamiprid and IM2 were 6.2 min and 5.6 min, respectively. The resulting findings are summarized in Table 4. During these studies, the target compounds were separated.

Cost and Time Performances: The total time for the analysis of a single sample was less than 20 min. For sequential analysis, a batch of 24 samples could be analyzed in less than 4 h. These findings became requirements for the routine assay. The short analytical time increased the sample throughput for analysis.
Application to Real Milk Samples: A total of 20 samples of commercial raw milk purchased from convenience stores in Osaka, Japan, were analyzed using the method. No samples contained detectable concentrations of acetamiprid and IM2. All chromatograms were free from interferences.
Conclusion
A simple sample preparation step followed by an HPLC method using an isocratic water mobile phase and PDA detection was successfully established for the simultaneous quantification of acetamiprid and IM2 in cow’s milk. The method validation data were well within the international method acceptance criteria. The procedure was easy to use, rapid, and space saving and resulted in high recovery and repeatability with considerable saving of analysis time and cost. The technique should be considered for use as an international harmonized method for determining acetamiprid and IM2 in milk.
References
- D. Cressey, Nature496, 408 (2013).
- European Commission, Regulation (EU) No 485/2013, (Brussels, Belgium, 2013). Available: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:139:0012:0026:EN:PDF
- European Commission, Bees & Pesticides: Commission goes ahead with plan to better protect bees (2013). Available: http://ec.europa.eu/food/animals/live_animals/bees_en
- European Food Safety Authority (EFSA), EFSA assesses potential link between two neonicotinoids and developmental neurotoxicity (2013). Available: http://www.efsa.europa.eu/en/press/news/131217.htm
- FAO, Acetamiprid (JMPR 2005). Available: http://www.fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Report11/Acetamiprid.pdf
- MRLs Database (The Japan Food Chemical Research Foundation). Available: http://www.m5.ws001.squarestart.ne.jp/foundation/search.html
- K.A. Ford and J.E. Casida, Chem. Res. Toxicol. 19, 944–951 (2006).
- G. Tanner and C. Czerwenka, J. Agr. Food Chem.59,12271–12277 (2011).
- European Food Safety Authority (EFSA), EFSA Journal9, 2328 (2011).
- K. Taira, K. Fujioka, and Y. Aoyama, Plos One8, 1–12 (2013).
- T. Yamamuro, H. Ohta, M. Aoyama, and D. Watanabe, J. Chromatogr B969, 85–94 (2014).
- M. Gbylik-Sikorska, T. Sniegocki, and A. Posyniak, J. Chromatogr B990, 132–140 (2015).
- P.T. Anastas and J.C. Warner, Green Chemistry: Theory and Practice (Oxford University Press, Oxford, United Kingdom, 1998).
- EU classification (The Dangerous Substances Directive 67/548/EEC): Council Directive 67/548/EEC of 27 June 1967 on the approximation of laws, regulations and administrative provisions relating to the classification, packaging and labelling of dangerous substances. Available: http://eur-lex.europa.eu/eli/dir/1967/548/oj
- FDA/CDER/CVM, Guidelines for Industry – Bioanalytical Method Validation (2001). Available: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf
- US Food and Drug Administration, Reviewer Guide,
- Validation of Chromatographic Method Center Drug Evaluation and Research (CFDER) (FDA, Silver Spring, MD, USA,1994).
- International Conference on Harmonization, Work Products, ICH Guidelines, Quality Guidelines (ICH, Geneva, Switzerland) Available: http://www.ich.org/products/guidelines.html
- AOAC International, Method Validation, Guidelines, & References. Available: http://www.aoac.org/vmeth/guidelines.htm
- Codex alimentarius commission, ALINORM 01/23, Codex Committee on Methods of Analysis and Sampling, Joint FAO/WHO Food Standards Programme, the 23nd Seccion of the Codex Committee on Method of Analysis and Sampling. Available: http://www.codexalimentarius.net/web/archives.jsp?year=01
- GL Sciences, Monolithic SPE Column for the Purification and Enrichment of Small Amount Sample, MonoSpin Series. Available: http://www.glsciences.com/products/monolithic_products/mono_spin.pdf
Naoto Furusawa is an associate professor at the Graduate School of Human Life Science, Osaka City University, Japan. He is an expert in multiresidue analyses. He has published more than 90 research papers in international journals. He received his Ph.D. in 1995.












Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.



Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

