What Exactly Are Raw Data?


Raw data is a term that is used in both good manufacturing practice (GMP) and good laboratory practice (GLP) laboratories but it can create misunderstanding. What exactly does raw data mean and what electronic records are within the scope of the term?
R.D. McDowall, R.D. McDowall Ltd, Bromley, Kent, UK
Raw data is a term that is used in both good manufacturing practice (GMP) and good laboratory practice (GLP) laboratories but it can create misunderstanding. What exactly does raw data mean and what electronic records are within the scope of the term?
Readers of this column will know that the definition of raw data is an interest of mine and has been discussed several times over the last 20 years. Here, we discuss it once more and it is déjà vu all over again. If you want to start a scientific argument with colleagues working in a regulated laboratory you can always ask the question: what exactly are raw data? The answer depends on which side of the good laboratory practice (GLP) or good manufacturing practice (GMP) fence you happen to be sitting on:
- “Original observations” will be the GLP answer
- “Raw data are used to create other records” is the GMP response.
The problem is that the term raw data is not always fully understood and can lead to regulatory nonâcompliance. This is compounded by the failure of regulatory bodies to define the term or to provide guidance. Therefore, we will explore what raw data actually means for a chromatography data system (CDS) in today’s regulated GLP and GMP environment.
In the Beginning
Raw data was first defined in the FDA’s Good Laboratory Practice regulations in 1978 (1) and was described in 21 CFR 58.3(k) as:
Raw data means any laboratory worksheets, records, memoranda, notes, or exact copies thereof, that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study.
There are other elements in the definition but this is the important part. In the context of chromatographic analysis, the focus is only the file generated from an injection as the raw data (original observation), and the metadata used to collect it and also any further interpretation of the data file is ignored. From this misconception, the current problems that we have with data integrity begin.
Most chromatographers working in regulated laboratories have never read the regulations. They have been interpreted for them by the great and the good and then handed down like tablets of stone to them to implement and follow. For the raw data debate, the training emphasizes that original observations must be captured, secured, and protected.
BUT...this is the time to use one of the corollary’s of Murphy’s Law, Cahn’s Axiom, which states:
When all else fails read the manual, SOP, or regulation.
Look back to the definition of raw data. Yes, it talks about original observations. But there is more, much more.
Raw data means … records …. that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study.
Do you see the problems?
It is not just original observations but also “activities”. There are now many more records that could be included. However, the killer line is the one everybody forgets: necessary for the reconstruction and evaluation of the report for that study. We’ll return to this topic after discussing raw data from a GMP perspective.
Later, Much Later in Europe
Let us move the clock forwards to 2011, when the European Union issued an updated version of EU GMP Chapter 4 (2). Chapter 4 details the main requirements for documentation in a single location and can be quite explicit in expectations for specifications, instructions, and records. In the principle of the chapter, under the subject of records, we have the following regulatory requirements for records (2):
- Provide evidence of various actions taken to demonstrate compliance with instructions, e.g. ….. manufactured batches a history of each batch of product…
- Records include the raw data which is used to generate other records.
- For electronic records, regulated users should define which data are to be used as raw data.
- At least, all data on which quality decisions are based should be defined as raw data.
OK, trivia time!
Q: Where is the definition of raw data contained in the EU GMP?
A: Nowhere!
Q: What is the definition of raw data in the US GMP?
A: There isn’t one!
The main problem is that when a new term is introduced into a regulated environment there should be a definition of that term so that organizations can interpret and apply it. However, EU regulators have failed to provide the definition of raw data to enable industry to begin any interpretation.
Also, you’ll note that the fourth bullet point contains the ever popular “at least” phrase. How do inspectors interpret this? This is
the minimum but we would expect more. How does the industry interpret this phrase? This is all we will do. Life is beautiful (at least for consultants).
FDA to the Rescue?
We discussed the FDA’s definition of raw data that dates from 1978 earlier, but now the agency has recently contributed to the raw data debate by issuing a proposal for updating GLP (3) based on a quality system approach-perhaps the working title of GLP Quality System is a bit of a clue? One of the aims of the revised regulation is to address the impact of computerized systems. The proposal has a modified raw data definition that addresses copying requirements, computerized systems, and includes the pathology report. It reads:
Raw data means all original nonclinical laboratory study records and documentation or exact copies that maintain the original intent and meaning and are made according to the person’s certified copy procedures.
Raw data includes any laboratory worksheets, correspondence, notes, and other documentation (regardless of capture medium) that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study.
Raw data also includes the signed and dated pathology report (3).
What the agency has done, by adding other documentation regardless of capture medium and copying, is to eliminate the outdated record examples in the original definition. The specific inclusion of “the signed and dated pathology report” to what is considered as raw data changes the definition of raw data from mere “original observations” to emphasize the whole process-from analysis to reporting is now included under the term raw data.
Extracting Principles for GXP Raw Data
What does this mean in practice? How can we interpret raw data for both GLP and GMP laboratories? Let us look at either the proposed or current GLP definition of raw data.
We will begin with original observations. How do we make the original observations for a chromatographic analysis? We need:
- A sampling plan (GMP) or study protocol (GLP) that documents how samples will be taken, stored, and transported.
- Sample(s) with the relevant information, such as identity, study, batch, or lot number, analysis request, etc.
- A qualified chromatograph and validated CDS
- An appropriate and validated method, including the preparation instructions for the sample presentation to the instrument.
- Reference standards
- Qualified staff to perform the work.
From these prerequisites, the analysis is undertaken and one or more data files will be generated and saved by the system. These are the first part of the raw data. No, not just the data files themselves, but all the other associated contextual metadata that must be linked together to support the generated files containing the identity of the instrument, acquisition method sequence file, instrument control file and the analyst performing the work, date and time stamps on the files, audit trail entries, and so on, as shown in Figure 1.


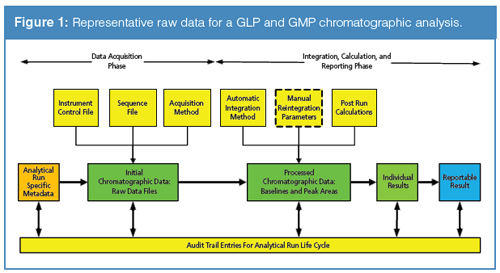
An Interlude for Recap
Remember the definition of raw data as “original observations”? The data files plus the contextual metadata described above must be considered as part of the raw data because they are used to acquire the original observations. However, there are “activities” to consider as a part of raw data, as we shall discover now.
Continuing the Raw Data Journey
As we have only acquired the original chromatography data files, we now need to interpret the data in accordance with the analytical procedure that we are using. In doing so, we will generate more records of the work that would include laboratory notebook entries and completed blank or template forms (4), together with further contextual metadata, such as integration of the peaks, calibration curve used, identity of the library or reference standards, the person who performed the work, date and time of work, associated audit trail entries, and so on.
At the completion of the analytical work, a draft report can be generated for review by a second person reviewer. This may result in changes as a result of typographical errors or misinterpretation of a spectrum, which will result in more metadata and possibly more files being created. Moreover, any interpretation, calculations, or transformations must be transparent, traceable, and understandable. At the end of the process, a final approved report or Certificate of Analysis (COA) is available. From the new FDA definition (3), this report is explicitly part of the raw data. However, implicitly the report is part of the raw data under the current GLP definition (1) that nobody bothers to read.
Now we have a better understanding of what constitutes raw data-all records, including any contextual metadata, generated from the sample to the report. Simple!
Visualizing What Raw Data Are
Figure 1 demonstrates the scope of raw data and is derived from a recent Questions of Quality instalment (5). The raw data trail starts with the sample preparation step with work documented in laboratory notebooks and worksheets for which there is accountability (4,6,7) before the sample is presented to the instrument. There are data files for controlling the instrument, acquiring the data, and identifying the samples. Next, the files are integrated and then interpreted as required by the method and the reportable result calculated and reported. As noted above in the GLP definitions of raw data (1,3), the report itself is part of the raw data for the work.
Summary: Raw Data = Complete Data
In this column I have discussed that raw data is more than just original observations. The term encompasses all records that have been created, from sampling to reporting, and highlights that all stages of the process should be transparent and understandable. It also means that an auditor or reviewer can track back from a result in the report to the original observations, or forward from the sample to a result in the report.
What should also be apparent to you is the similarity between raw data in a GLP context and complete data for GMP as per 21 CFR 211.194(a) (8). In my view the two terms are equivalent and mean the same thing, regardless of the GXP discipline that one is working to. Quod erat demonstrandum?
Acknowledgements
I would like to thank Lorrie Schuessler for helpful comments made while reviewing this article.
References
- Food and Drug Administration, 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies (FDA, Washington, DC, USA, 1978).
- European Commission Health and Consumers Directorate-General, EudraLex: Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation, E. Commission, Ed. (Brussels, Belgium, 2011).
- FDA, “21 CFR Parts 16 and 58 Good Laboratory Practice for Nonclinical Laboratory Studies”, proposed, Federal Register 81(164), 58342–58380 (2016).
- C. Burgess and R.D. McDowall, LCGC Europe29(9), 498–504 (2016).
- C. Burgess and R.D. McDowall, LCGC Europe28(11), 621–626 (2015).
- Food and Drug Administration, Inspection of Pharmaceutical Quality Control Laboratories (FDA, Rockville, Maryland, USA 1993).
- Food and Drug Administration, Draft Guidance for Industry Data Integrity and Compliance with cGMP (FDA, Silver Spring, Maryland, USA, 2016).
- Food and Drug Administration, 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (FDA, Silver Spring, Maryland, USA, 2008).
“Questions of Quality” editor Bob McDowall is Director at R.D. McDowall Ltd., Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column should be addressed to the editorâin-chief, Alasdair Matheson, at alasdair.matheson@ubm.com








What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






