Rapid Method Development for Industrial LC Separations Using Automated Screening of Stationary Phases and Solvents
Special Issues
Rapid Method Development for Industrial LC Separations Using Automated Screening of Stationary Phases and Solvents.
High performance liquid chromatography (HPLC) is an important separation technique that is used in research, development, and quality control of many chemical, agricultural, biochemical, and pharmaceutical products and intermediates. Development of new HPLC methods is a common task for the experienced chromatographer. Drivers for method development include development of new products, changes in registration for products or product formulations, new requirements for better and faster separations, and finally changes in the availability of certain LC columns as a result of new product developments or discontinuation of existing products. For these method development efforts, a wide range of columns, solvents, and other experimental parameters are used to develop a separation based upon the chemistry of different molecules (1).



Matthias Pursch
In the last decade, many column and instrument developments have occurred that allow users to take advantage of higher separation efficiencies per column length. Part of this technology has been due to a continuous reduction of column particle size from the traditional 5–10 µm to a range of 1.5–3.5 µm (2). In addition, superficially porous materials have been introduced (2.6–2.7 µm particle size), which provide comparable peak capacity of sub-2-µm particle columns, but at about only half the column back pressure (3,4). Also, the effect of particle size distribution of the base silica on column back pressure has received more attention (5), and efforts currently are being undertaken by various vendors to improve the efficiency/back pressure ratio. All of these developments are important milestones for fast or high-resolution separations. However, as shown in equation 1, not only column efficiency N, but selectivity α and capacity factor k are required to obtain the optimum resolution Rs:
Rs = 0.25 N0.5 (α–1/α) (k2/1 + k2) [1]
Improving the selectivity α is often less straightforward than improving the number of theoretical plates N or altering retention k. For this reason, development of new LC methods often does not go beyond the use of C8 or C18 columns. To take advantage of selectivity, a wider variety of available stationary phases should to be assessed. These could include specific polymer-based stationary phases, and, in particular, some of the many silica reversed-phase columns, such as C3, C8, C18 (with different types of endcapping), polar-embedded C18, C30, aromatic phases (phenyl, phenyl hexyl, biphenyl, diphenyl, naphthyl), cyanopropyl, or even fluorinated phases.
Phase-optimized LC has been introduced to take advantage of optimal selectivity by coupling column segments packed with different stationary phases (6,7). This effort is supported by software (6) for isocratic separations and can suggest different lengths of different stationary phase segments to obtain the optimum selectivity. While this innovative concept allows the best column to be used for a specific separation problem, the individual column segments are not available for 3-µm or sub-2-µm particles. Thus, fast or high-resolution LC separations are, with some exceptions, difficult to achieve. It should be noted that gradient separations are now supported by a software algorithm (8), which should further increase the utility of this novel approach.
An alternative approach to finding the best separation selectivity is to broadly screen various types of column chemistries that are available from various column manufacturers. This approach can become a very tedious process if the column-selection process is performed manually. For this reason, using a semiautomated system that is capable of sequentially screening different columns and different mobile-phase gradients would provide a substantial productivity increase. Valve-based solutions are facilitating such screening programs and have become available on the market.
For our work, the screening efforts were executed using the Model 1200 RRLC (HPLC) and 1290 Infinity LC Method Development Solution (ultrahigh pressure LC or UHPLC) systems provided by Agilent Technologies (Santa Clara, California). The HPLC and UHPLC systems consisted of a binary pump with attached solvent selection valve, a high-performance autosampler, two connected column compartments, and a diode-array detector. Chemstation software was used for instrument control and data analysis. The method-development system typically has a series of up to eight columns installed to allow for a broad stationary phase screening. Additional valve switching can be applied to screen between several organic modifiers (for example, acetonitrile, methanol, ethanol, isopropanol, and so forth) or different buffer characteristics (for example, buffer type, buffer strength, and buffer pH). The screening experiments are set-up using dedicated method development software provided by Agilent. In the present work, both short and long columns (that is, 50-mm and 150-mm lengths) with different particle sizes (1.7 µm, 1.8 µm, and 2.7 µm) packed with different stationary phases with different chemistries were utilized for screening efforts for fast or high-resolution LC separations. Four examples of method development for the chemical and agricultural areas are presented in this article.
Impurity Profiling of Herbicides
The first example details the application of column and solvent screening for impurity profiling of herbicides. For the automated set-up, six different sub-2-µm columns (50 mm x 4.6 mm) were used. The range of stationary phases investigated included endcapped and nonendcapped C18 silicas, as well as cyanopropyl and phenyl hexyl columns. The Bonus-RP phase (Agilent Technologies) is an alkyl stationary phase that contains a polar embedded group. The Extend C18 (Agilent Technologies) is based upon bidentate alkylsilanes, making the bonded silica more stable at higher pH.
Changes in selectivity were readily observed for columns with different surface chemistries. A gradient screen for all six columns with acidified aqueous and two organic modifiers was used to identify the phenyl hexyl column as the stationary phase providing the best selectivity with both acetonitrile and methanol gradients as shown in Figure 1. Noteworthy is its ability to separate component A very nicely from the main peak, while still providing sufficient separation between components B and C.


Figure 1
For the initial screening effort, a very broad gradient is recommended to ensure that the components of interest are actually eluting off the respective columns but also show retention. This broad gradient also accounts for differences in column chemistries (for example, carbon load and stationary phase polarity). Although not used in the present example, the same "broad-based" approach could be used for scouting different gradients and different operating temperatures. When no conditions from an existing separation are available, a prescouting run under standard conditions — for example, 5–95% acetonitrile in water gradient on a C18 phase or other columns at 40 °C — can be used to obtain a reference. From here, the common rules for modifying retention can be applied to set boundaries on the gradient composition. The actual mobile phase pH (buffer composition) should be dictated by knowledge of the analyte chemistry.
Also, for a new screening campaign, a blank screening gradient is strongly recommended for each stationary phase to wash off potential contaminants that might be present on the respective LC columns or observed as contaminants in the solvents themselves. In terms of separation efficiency, one has to consider the additional dead volume that is introduced with the additional two valves. Using 4.6-mm i.d. columns, this effect is usually negligible. In our series of experiments, about 12,000 plates were obtained for the 50 mm x 4.6 mm columns under isocratic conditions. This corresponds to the theoretically expected column efficiency. For 2.1-mm i.d. columns, a decrease of separation efficiency compared to operation without valves needs to be taken into account. In most gradient experiments though, analytes are refocused at the head of the column under the low %B starting conditions and the impact of band broadening in additional tubing and valves before the column is eliminated, leaving only the influence of dead volume between column and detector, which can be optimized by judicious selection of tubing internal diameter and length and the detector flow cell volume.
Trace Level Target Compounds in Aromatic Amine Oligomer Mixture
The second example is related to the separation of trace-level target compounds (TC) in an aromatic amine oligomer mixture. We first investigated manual method development using an acetonitrile–acetate buffer mobile phase and a Fused Core Ascentis Express C18 column (Supelco/Sigma Aldrich, Bellefonte, Pennsylvania). The column was 150 mm in length to provide sufficient peak capacity for separation of the respective oligomers. Under these conditions, various isomers (present at low concentration) of the oligomers were well separated, but overlapping with the target compounds. Optimization was performed through numerous iterations of gradient conditions and temperature in an attempt to find suitable optimized conditions. The final separation, which was obtained after about three weeks of intensive efforts by experienced method development chemists, is shown in Figure 2. While the overall separation is excellent, it also possesses a few constraints. TC 1 is just barely separated from another sample constituent. TC 2 is eluted on the tail of a very large peak, which would make quantitation difficult for very low peak heights. Also, it was not clear whether overlap with another component occurred for TC 2. Finally, the final method was found to be very sensitive to slight changes in mobile-phase composition.


Figure 2
Therefore, it was decided to also apply automated screening of various stationary phases in order to explore alternative column selectivities. As a result, two columns — a phenyl hexyl ( example not shown here) and especially a cyanopropyl column — provided better selectivities than the C18 column. Chromatograms are illustrated in Figure 3. It is apparent that the target compounds "moved" to earlier elution times relative to the main sample components for the cyanopropyl column versus the C18 stationary phase. The inset in Figure 3 shows that the target compounds are well separated from other sample constituents using an acetonitrile–buffer mobile phase gradient. In this case TC 1 is eluted on the tail of a large peak (effect seen at low peak heights); this was found to be acceptable for this separation problem, because TC 2 is well separated from other sample constituents. Fine tuning of the optimized column–solvent combination required a minimal amount of time, because the improved selectivity provided more space for successful separation of the target components without overlap. This screening effort (columns and methanol–acetonitrile as organic modifiers) was carried out overnight and yielded the optimal separation after some additional gradient adjustments within two days. Compared to the "manual" method development, which took about three weeks, this was a very significant productivity increase.


Figure 3
High-Resolution Separation of Insecticides
The third example deals with high-resolution separations, which are becoming of greater importance in the HPLC area, in particular for very complex sample mixtures. Examples of highly complex samples include peptide digests, isomeric and oligomeric samples (9) (as shown in Figure 2), or natural products. These samples require high peak capacity columns — for example, 150-mm-long columns packed with either sub-2-µm (Zorbax StableBond-C18, Agilent or Acquity C18, Waters, Milford, Massachusetts), or superficially porous silica particles (Halo, Advanced Material Technology, Wilmington, Delaware), each of which provide up to 35,000–40,000 plates for an isocratic separation. A semisynthetic mixture of insecticides was used for high-resolution C18 column screening as illustrated in Figure 4. About 30–35 peaks are counted in the chromatograms, some of them only partially resolved. In the figure, it is apparent that these columns, although all of them are C18, produce quite different selectivities and retention factors k, in particular, for some of the early-eluted components. The superficially porous column was found to provide better overall separation in the first half of the chromatogram, while the other two columns yielded better resolution in the vicinity of the two large peaks. Overall peak capacity for these columns is very similar. Therefore, depending upon the requirements of the final separation, one of these columns could be selected for further gradient optimization or exploration of additional organic modifiers. No further investigation of additional optimization was performed.


Figure 4
We would like to discuss briefly the use of additional organic modifiers for gradient optimization. With the recent acetonitrile crisis, the search for alternative organic modifiers has resurfaced. While methanol most often is considered as the replacement for acetonitrile, it does not always provide the optimal selectivity. Ethanol has been propagated recently as a solvent for "green chromatography" in hydrophilic interaction liquid chromatography (HILIC) (10). It is attractive because of its different selectivity and also its biodegradability, which will affect waste disposal. However, because of its high viscosity and corresponding high back pressure, ethanol has been used rarely for HPLC. The same applies for 2-propanol, which has even higher viscosity than ethanol. However, with the availability of UHPLC instrumentation (800–1200 bar), the screening of these high-viscosity organic modifiers is facilitated greatly. In the next example, we will be investigating solvent selectivity by the using these less common organic modifiers.
Rapid Column and Solvent Screening for Nitroaromatic Compounds
The final example is provided in Figure 5 for the separation of nine nitro aromatic compounds. Here, the desire was to find a suitable separation that does not require the use of acetonitrile. Using a 1290 Infinity LC system and 50 mm x 4.6 mm sub-2-µm columns, automated screening of six columns and four organic modifiers was executed. The whole campaign consisted of 48 experiments (six columns, four mobile phases, two injections per column, blank and sample runs) and took less than 6 h, thus, the results could be reviewed during the day. As expected, the chromatograms displayed in Figure 5 show significant differences among the columns and solvents used. Figure 6 shows the effect of organic modifier for a particular C18 column. While ethanol and 2-propanol were not able to resolve all nine components of interest, it was found that methanol is a suitable solvent for replacement of acetonitrile for these nitro aromatic species.


Figure 5
Table I lists the dynamic viscosities of various organic modifiers and the related maximum column back pressures observed during the gradient separations. For each column and each solvent, the highest back pressure during the separations was determined. It should be noted that the maximum pressure is at different compositions for each solvent. As expected, the lowest values are always obtained for the superficially porous particle column, in this case the C8 column. Higher back pressures are observed for sub-2-µm columns ("Sub-2-µm A"). The highest values ("Sub-2-µm B") represent a sub-2-µm column that had been used extensively for other separations before this screening campaign, and it did show signs of partial plugging. The data in Table I show why acetonitrile is such a popular solvent. In addition to its excellent elution properties, it produces very low column back pressures on reversed-phase systems. Back pressures of up to 630 bar were measured for ethanol and up to 840 bar for 2-propanol using the 50-mm columns, thus exceeding the ranges of conventional LC and even moderate UHPLC instrumentation. While for the present case study ethanol and 2-propanol solvents did not produce optimal selectivity, they should become more important in future method development efforts of other compound classes.

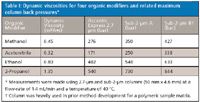
Table I: Dynamic viscosities for four organic modifiers and related maximum column back pressures*
Conclusion
Several case studies for separations of pesticides and chemical intermediates were presented to demonstrate the power of automated column and solvent screening for rapid LC method development. For some examples, rather unusual column chemistries such as cyanopropyl or phenyl hexyl provided the optimal selectivity for a particular separation. The success of these surface chemistries can be reasoned by providing π-π-interactions with the analytes (11).

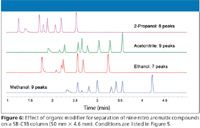
Figure 6
Using automated LC method development with very efficient columns such as sub-2-µm or superficially porous silica materials, the time needed for an optimal separation can be reduced to several days or less, compared to several weeks if all experiments were done manually. In some cases, the optimal method was already obtained within a few hours after one full screening campaign. In other examples, the initial screening results were of sufficient quality so that additional gradient screening was able to provide the final separation rather quickly. Thus semiautomated method development will provide substantial productivity enhancement for today's analytical laboratories.
Acknowledgment
The authors would like to thank Angelika Gratzfeld-Huesgen, Michael Frank and Helmut Schulenberg-Schell (Agilent Technologies) for excellent support. Dave Albers (The Dow Chemical Company) is acknowledged for helpful discussions.
Matthias Pursch*, Rob Edam*† , and Freddy Van Damme†
*Dow Deutschland GmbH & OHG, Analytical Technologies, Rheinmuenster, Germany.
† Dow Benelux BV, Analytical Sciences, Terneuzen, Netherlands.
Direct correspondence to: mpursch@dow.com.
Matthias Pursch is global LC technology leader at The Dow Chemical Company. His current research interests include development and identification of new technology to advance liquid phase separations for a wide range of chemical and agricultural applications. Matthias Pursch is author/co-author of about 40 publications.
References
(1) L.R. Snyder, J.L. Glajch, and J.J. Kirkland, Practical HPLC Method Development, 2nd ed. (Wiley-Interscience: New York, 1997).
(2) R.E. Majors, LCGC 27(11), 956–972 (2007).
(3) J.J. Kirkland, T. Langlois, and J.J. De Stefano, Am. Lab. 39, 18–21 (2007).
(4) P. Yang, G.R. Litwinski, M. Pursch, T. McCabe, and K.J. Kuppannan, Sep. Sci. 32, 1816–1822 (2009).
(5) D. Cabooter, J. Billen, H. Terryn, F. Lynen, P. Sandra, and G. Desmet, J. Chromatogr., A 1204, 1–10 (2008).
(6) S. Lamotte, GIT Spezial Sep. 28, 52–53 (2008).
(7) M. Kuehnle, J. Rehbein, K. Holtin, B. Dietrich, M. Gradl, H. Yeman, and K. Albert, J. Sep. Sci. 31, 1655–1661 (2008).
(8) M. De Beer, F. Lynen, K. Chen, P. Ferguson, M. Hanna-Brown, and P. Sandra, Anal. Chem., in press.
(9) M. Pursch, A. Schweizer-Theobaldt, H.J. Cortes, A. Gratzfeld-Huesgen, H. Schulenberg-Schell, and B. Hoffmann, LCGC Eur. 21, 152–159 (2008).
(10) A. Dos Santos Pereira, F. David, G. Vanhoenacker, and P. Sandra, J. Sep. Sci. 32, 2001–2007 (2009).
(11) D.H. Marchand, K. Croes, J.W. Dolan, L.R. Snyder, R.A. Henry, K.M.R. Kallury, S. Waite, and P.W. Carr, J. Chromatogr., A 1062, 65–78 (2005).



















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


