Practical Guidelines in the Care and Maintenance of Capillary GC Columns
LCGC Europe


Modern gas chromatography (GC) capillary columns are rugged and forgiving but some care should be taken when handling and using these high-efficiency columns. In this instalment of "Column Watch", the authors discuss the issues surrounding avoiding column breakage, stationary phase damage and column contamination. Prolonging the life of a column by keeping an oxygen-free system, providing a cleaner sample and not exceeding the upper temperature limit of the stationary phase are highlighted in this practical discussion.
Modern gas chromatography (GC) capillary columns are rugged and forgiving but some care should be taken when handling and using these high-efficiency columns. In this instalment of "Column Watch", the authors discuss the issues surrounding avoiding column breakage, stationary phase damage and column contamination. Prolonging the life of a column by keeping an oxygen-free system, providing a cleaner sample and not exceeding the upper temperature limit of the stationary phase are highlighted in this practical discussion.
Many gas chromatographers rely on their gas chromatography (GC) system to function properly under extreme and demanding conditions that put their system and column under heavy stresses. The column, a simple fused-silica open-tube capillary, is often held responsible when poor chromatography arises and yet it is never praised for the hard work that it does on a daily basis. These columns have fallen victim to the high-throughput analysis that is common in today's laboratories. Often, little regard is given to what is injected onto the column. When analysts change columns or perform routine maintenance on a system, the columns are sometimes mishandled. Many chromatographers believe that columns have become indestructible and treat them as such. While technological advances have been made in column manufacturing, care should still be taken when handling or using a capillary column.
Even if your GC instrument is used in relatively "light duty" situations (for example, very clean sample matrices, few samples run per day) there are things that you can do to ensure that the column and the GC system as a whole perform at an optimum. We will discuss some precautions that will help prolong the life of capillary columns, thus, minimizing downtime and increasing productivity.
Column Breakage
Many chromatographers have one of two perspectives when it comes to capillary GC columns. Some think that they are nearly indestructible and that a 0.53 mm i.d. column can handle a bending radius of a finger. Others feel that the capillary column should be treated as if it were a fine piece of crystal. The correct perspective is somewhere in between. The reality is that the column is very durable and robust but still needs to be handled with care.
The capillary column is a fused-silica "pure quartz" open tube that is coated on the outside with a polyimide polymer and on the inside with a liquid or solid stationary phase. The capillary tube, being constructed of quartz, is actually very strong on its own. The polyimide coating provides the outer surface with a layer of protection against abrasions that could scar the tube and eventually lead to column breakage. It also protects the column from any surface flaws that if exposed to the elements in the oven (heat and moisture) would grow at an accelerated rate and lead to a failure (breakage). As the diameter of the column cage winding decreases, the stresses on the fused-silica tube increase. This observation is equally true for an increase of the inside diameter of the capillary tubing. The minimum bending or cage radius increases with an increase in column diameter. Some chromatographers try to determine how well a column will perform by bending the column to an extremely small radius, thus, testing its brittleness. If the column breaks at a larger bending diameter than what they are used to, the column is assumed to be "bad". How tightly you can bend a column has no bearing on how well it will perform chromatographically. The performance of the column depends upon the stationary phase inside the column, not the strength of the capillary itself.
The protective coating on the column helps it withstand breakage from its environment. If the integrity of this coating is compromised, so is the strength of the column. When you trim a column, you score the polyimide coating and slightly nick the quartz tube, creating an imperfection, at which point the column breaks freely. The column hangers inside a GC oven help keep the column out of contact with any surfaces that could potentially scar the polyimide coating and expose the quartz column. As the GC oven goes through heating and cooling cycles, the column vibrates a little, and over time, this exposed area could lead to a break.
A broken column is not necessarily a fatal event. If the break occurs within a metre or two of one of the ends, discarding that short portion of the column, trimming it to make sure that the end of the capillary is perfectly flat and reinstalling it will cause minimal differences in performance, if any at all. Adjusting your data station for the difference in column length will help retain the appropriate flow parameters for the analysis, and resolution will not change noticeably. If a break occurs in the middle of the column, rather than discarding a column that might still be good, try using a column union to rejoin the pieces. A union such as a press-fit connector can be used to join the two sides of the column together quickly and inexpensively. Again, because the break will probably not be clean, trim the ends of both sections of the capillary column before rejoining. There are other column connectors available if you are wary of the press-fit option. The number of unions down the length of a capillary column should be limited to two or three. The larger the number of unions, the more dead volume there is, which can lead to peak tailing. A union will be a less expensive alternative to replacing the entire column and your chromatography will generally not suffer as much as you might think.
The easiest way to avoid column breakage is to use good common sense and treat your column with care. Take care when installing or removing a column and try not to scrape it with or against any object that could potentially weaken the polyimide coating. When storing a column, seal the ends using an old septum and place it back in its original box instead of putting it unprotected in a drawer or on a shelf where it can come into contact with sharp objects. Using care and common sense will help keep the column in good shape for a longer period of time.
Stationary Phase Damage
The root cause of stationary phase damage is often difficult to determine because the symptoms are usually similar. The symptoms of stationary phase damage or degradation are often indicated by excessive column bleed, a higher degree of column activity, a loss of efficiency or a combination of these effects. The three main causes of stationary phase failure are prolonged exposure to oxygen, sample contamination and exceeding the upper temperature limit of the column.
Prolonged oxygen exposure: Stationary phase damage as a result of prolonged exposure to oxygen is the most destructive and the most common mode of column failure.1 Oxygen exposure primarily is because of the presence of a leak in the system. Leaks usually occur at the inlet and are caused by a loose septum nut or a bad septum. Sometimes leaks can be found in the tubing and connections after the gas traps have been replaced. Stationary phase damage because of oxygen in the column begins just above room temperature and the severity increases with increasing temperatures. Normal injections of small volumes of air or samples that contain air are not of concern, but prolonged exposure of oxygen is especially detrimental to a column.
One of the early signs of stationary phase damage will be an increase in the baseline signal at low temperatures. Normal column bleed occurs at temperatures 30–40 °C below the upper temperature limit of the column. Each stationary phase has its own upper temperature range. Not surprisingly, these temperature limits are related directly to the overall stability of the stationary phase. The higher the temperature limit, the more stable the phase. The amount of bleed produced by a given column is related directly to the type of stationary phase as well as the amount of phase present. Therefore, for a given stationary phase, the bleed will be higher for thicker films than for thinner films. The same also holds true for an increase in polarity or column diameter and length. These relationships are described by the graph of Figure 1. Column bleed is an equilibrium process, and as such, produces a rising baseline at elevated temperatures at which the stationary phase becomes less stable. Bleed produces neither discrete peaks nor an elevated baseline at low temperatures. Once you have introduced oxidative damage to the column, this "normal" baseline rise occurs at much lower temperatures, or might even be seen as an increased resting baseline under isothermal conditions.


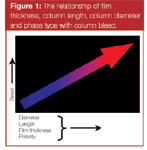
Figure 1
There are several ways to limit the amount of oxygen exposure to a GC system. The first is to make sure that you use a high grade of carrier gas. However, using the highest grade of gas from a supplier is never a guarantee that no oxygen is present in the cylinder. The next line of defence is to use the appropriate gas traps or purification system and to make sure that these are maintained or replaced on a regular basis. Initially, a large capacity trap followed by a smaller indicating oxygen trap ensures that oxygen is scrubbed from the carrier gas. An indicating trap that changes colour as its trapping efficiency decreases will notify you visually when your large capacity trap needs to be replaced before oxygen damage can begin to occur in your column. After you you are sure that your gas supply is oxygen free, a leak test on the column installation is always a good idea. Snoop (Swagelok, Solon, Ohio, USA), a commonly used leak detector, is never recommended to use for a leak test around a column. Because it is essentially a soap solution, components of that solution are semivolatile to nonvolatile and can cause problems within your column or instrument if it happens to get inside the flow stream. A 50:50 mixture of methanol or isopropanol and water is generally a safer solution used for leak testing around a column. The solution can be dispensed with a squeeze bottle. An alternative device to use is a leak detector available from GC accessory suppliers. These probes are very sensitive to trace amounts of carrier gas, feed gases for detectors such as the flame ionization detector or make up gases.
One of the best ways to be certain of a leak-free system is to inject an unretained compound. This is a compound that has no interaction with the stationary phase and, therefore, should be eluted as a very symmetrical peak. Any deviation in peak shape, such as tailing, is indicative of a poor column installation or a flow-path obstruction. Butane is an easily obtained inert compound, especially good for the flame ionization detector. All one has to have on hand is a disposable lighter.
Once you have established that you have a leak-free system, it is recommended you measure the average linear velocity of your carrier gas. GC systems calculate their flows based upon the dimensions of the column. Because the pressure needed to obtain a particular flow is dependent upon the length and internal diameter of the column, being accurate with any changes to those dimensions will ensure that the resolution remains constant, assuming that column damage has not occurred.
Sample-related degradation: A second contributor to column stationary phase degradation is sample related. Column technology has progressed through the years, and the stability of the stationary phases has increased during this time. Most stationary phases today are bonded and cross-linked. This characteristic increases the robustness of a column and provides significantly more resistance towards unfavourable injected solutes and solvents. A bonded and cross-linked column will resist damage to its stationary phase by water or organic solvents. Even with prolonged residence times of these solvents, the stationary phase will remain unharmed.
However, there are still certain solutes that should be avoided if at all possible. Samples to avoid are those that contain inorganic acids and bases such as KOH, NaOH, H2SO4, HNO3 and HCl to name a few. These are quite damaging to the stationary phase, even when neutralized. Occasionally, there might be small amounts of residual base or acid left in the sample. Of these, HCl will be the least damaging to a column under certain conditions. Because HCl is a gas at room temperature, it will have little to no residence time on the stationary phase itself, that is unless water is present. If water is present, the HCl will travel with the water through the column and will probably have an extended residence time in the stationary phase and begin to degrade it. If water is present during an isothermal run, temperatures above 100 °C will prevent the water from condensing on the stationary phase, and, as such, will also minimize any residence time that the HCl will have. If you are using a temperature-programmed run that starts at temperatures below 100 °C and HCl-acidified water is present, the front end of the column will be in the harshest environment. If damage occurs, the front end (1–2 m) can be trimmed to remove the affected area and hopefully restore the chromatography. For details on cutting or trimming a GC capillary column, consult reference 2.
As with oxygen damage, the symptoms of stationary phase damage initiated by chemicals are loss of resolution, peak broadening, peak tailing and a shift in retention time, to list a few. Damage by acids and bases not only affects the column, but they can cause interferences within the inlet as well. The solutions can "salt out" in the liner and react with samples before they even reach the column resulting in some of the same characteristics of stationary phase damage. This interaction will be particularly noticeable with acidic and basic compounds. Changing the liner and trimming the column by 1 m or so can eliminate the problems.
Exceeding upper temperature limit: A third facilitator to column phase degradation is a problem that is often overlooked. Exceeding the posted upper temperature limit for a column or maintaining a column at the programmed temperature limit for prolonged periods of time can speed up the degradation of the phase. Stationary phase bleed begins at 30–40 °C below the upper temperature of the column. Most columns have two upper temperatures reported on the box (see the example in Figure 2). The lower of the two is called the isothermal temperature limit, and the other is the programmed temperature limit (usually in parentheses or depicted to the right of the isothermal temperature limit). The latter is the temperature that the column can be exposed to for a short period of time (~15 min or so) that can occur during a temperature program.


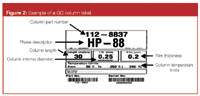
Figure 2
Sometimes when a new column is installed, or after maintenance with a reinstallation of the column, the carrier gas is left off accidentally or a different inlet is used on a dual-inlet system. Without the flow of a carrier gas, oxygen will not be purged from the column. As was described in the previous section, oxygen in the presence of elevated temperatures will cause stationary phase damage. Surprisingly, this is a common problem associated with elevated column temperature. The second most common failure as a result of higher column temperatures occurs when two columns with different stationary phases or with phases of different polarity are installed in the same GC oven. Typically, the higher the polarity of a column, the lower the stability of the stationary phase and the lower its upper temperature limits. One column can have an upper temperature limit of 260 °C, and the other of 325 °C. Upon conditioning, an analyst could inadvertently raise the temperature of the oven to 325 °C and damage the lower temperature column. Typical signs of thermal damage to a column will be similar to those of oxidative or chemical damage including poor peak shape, high baseline or excessive column bleed.
Column technology has changed over time and column manufacturers have produced more stable polar stationary phases that minimize some of the dual column set-up temperature mismatches. However, it is always a good practice to set the maximum column temperature in your method to the isothermal limit of the column. When two columns are installed in the GC system, always set all methods to the lowest isothermal temperature limit of the two columns.
Column Contamination
Contamination exists in even the cleanest samples and is routinely found in standards and even high-grade solvents. In many instances, there are more sample components that do not go through the column than do. Residuals exist to one degree or another in everything that is injected. Residuals are not always apparent. One simple way to check for residuals is to place 5–10 µL of the sample or standard onto a clean watch glass or microscope slide and place it on or near the hot injection port. After the solvent has evaporated, whatever is left behind on the glass is what will probably remain in the inlet or at the head of the column (see Figure 3).


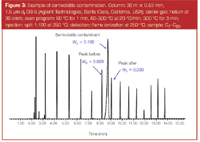
Figure 3
The two main classes of column contamination are nonvolatile and semivolatile. Nonvolatile contaminants are those that are essentially trapped either in the injector liner or near the front of the column and do not migrate under "normal" GC operating conditions. Semivolatile contaminates are more notorious and are those contaminants that migrate very slowly through the column and are eventually eluted. These are often the "big blobs" that are eluted unexpectedly and randomly from the column. However, such baseline upsets might also be a result of carryover from poorly optimized injection conditions. These extraneous peaks or baseline upsets might have been injected as part of a previous sample several hours or even days before. One way to identify semivolatile contamination is to examine the width of the offending peak compared with the peaks of interest. Very seldom will a very broad peak occur during the elution of a series of narrow peaks (see Figure 4). Besides the fact that the peak is not supposed to be there, if the so-called "bonus peak" is significantly wider than the peaks around it, this is an indicator that the material has been around for a while.



Figure 4
Contamination can mimic almost every type of chromatographic problem. Symptoms include ghost peaks, high bleed, sample decomposition, peak tailing or fronting and even changes in selectivity. Although we usually think of contamination as coming from samples, it can also come from several different sources namely split-vent traps, septa, ferrules, carrier gas, gas traps, sample vials or syringes.
The best way to eliminate contamination is to attempt to remove it before injection by employing clean-up steps during sample preparation. These steps can be as simple as passing the sample through a filter, performing sample extraction or the use of solid-phase extraction cartridges. Indeed, we find that the most fortunate chromatographers are those performing volatiles analysis by introducing their samples via purge and trap or headspace systems or by volatile analyte desorption from a suitable sorbent medium (for example, solid-phase microextraction, polydimethylsiloxane-coated stir bar and so forth). Columns used for these types of "clean" applications can last for years. These analyses lend themselves to longer column life because only the volatile materials are allowed onto the column, leaving any residues behind with the sample matrix. Even so, these ancillary injection techniques can be prone to gas leaks from an overused septum or worn valves and transfer line connectors, so attention must be paid to maintaining a leak-free gas system.
In the event that residues cannot be removed before injection, there are other techniques that can be employed. The first such method is to use glass wool placed at the lower end of the inlet liner. The glass wool will help to trap any nonvolatile material before the column. Another method is to use a guard column or retention gap, which is simply a 3–5 m piece of bare fused-silica capillary that is attached to the front of the column or is built-in as part of the front of the column. Any semivolatiles or nonvolatiles that make it past the glass wool will deposit on this section of column. When the time comes for inlet maintenance, a section of the guard column is removed, thus, extending the overall life of the analytical column.
Many GC problems are inlet-related or more accurately described as "front-end" related. As part of a normal inlet maintenance procedure, many times, only the liner is changed while the front end of the column is ignored. Presumably the liner is being changed because of known contamination problems. Because of the close proximity of the head of the column to the inlet liner, it is a good practice to always trim 10–20 cm off the column in conjunction with changing the inlet liner. For electronic pneumatic controlled GC systems, after trimming the front end of the GC column, a commonly overlooked step is to update the length of the column in the software. This will keep your column flows consistent because the carrier gas flow provided by the instrument relies on this dimensional information. Informing the GC software of this change will keep your retention times from shifting slightly after the column is trimmed.
After you have determined that there is a contamination problem, the simplest remedy is to trim a portion of the front of the column. Depending upon the severity of the problem, as much as 1 m or more might need to be removed. Normally, on a column longer than 20 m, this operation will not have a major impact on resolution. However, if trimming the column does not restore the performance, then the contamination is not localized to the front of the column. The contamination can reside in other parts of the inlet such as the liner or inlet body. The contamination can exist throughout the entire length of the column. When this form of contamination occurs, many analysts immediately
bake-out the column by heating it to its upper temperature limit for a period of time. A prolonged bake-out is tempting because of its simplicity; however, it is the least effective way to remove contaminates and can cause more problems than it solves. Baking out the column can cause contaminants to irreversibly polymerize or caramelize on the stationary phase, thus, permanently damaging the column.
A better solution is to rinse the column with solvent. Although the rinse procedure is more involved than a simple bake-out, the procedure is still relatively simple and definitely more effective. Solvent rinsing can be performed on any bonded and cross-linked phase only. Nonbonded phases cannot be rinsed. If there is any question as to whether your column can be rinsed, contact the manufacturer to be sure. Rinse kits are available for purchase or can be homemade (see Figure 5). Solvents with varying polarities should be used to remove the widest range of contaminants. Rinse in the order of most polar to least polar. Each successive solvent must be soluble in the previous one. The injection solvent should also be included when possible. A good general-purpose solvent list might be methanol, methylene chloride, and hexane. If aqueous-based samples have been injected or salts are suspected as contaminates, include water as one of the rinse solvents. Try to avoid any high boiling solvents. Anywhere from 5 to 10 mL of each solvent should be used, performing the rinse from the detector end to the injector end. This reversing of the column is recommended strongly, because most contaminants are likely in the front part of the column and the removal path is shorter for solubilized contaminants.



Figure 5
Be sure to "dry" the column phase thoroughly by purging with nitrogen for ~30 min after rinsing before placing the column back in the GC oven. Install the column in the injector side only and pass carrier gas through the column for 10–20 min, then install the column on the detector side and check for leaks. Start the oven program at 40 °C and ramp the temperature at 2–3 °C/min to the isothermal maximum. Hold this temperature until the baseline stabilizes, which should take only 30–60 min. All of these steps are meant to ensure that no residual rinsing solvent is left behind in the stationary phase. If solvent remains and the column is placed in the oven and heated too quickly, the rapid vapour expansion could cause damage to the internal polymeric layer.
Conclusion
While capillary columns are not as fragile as a piece of crystal, they are also not indestructible. Care should be taken to ensure the robustness and productivity that they can provide. Maintenance requires some periodic downtime. Minimizing this downtime is pertinent to the high sample throughput labs of today. Maintenance is always less time consuming than repair primarily because maintenance is a scheduled event that when properly implemented, will always be less timely than unexpected column, injection-port or detector failures caused by neglect. Topics that we discussed earlier, such as handling the column more gently, taking an extra moment to clean up a sample or taking an extra second to double check the instrument method parameters before starting the system, is critical to extending the life of a column. It is highly recommended that more frequent replacement of inlet liners will help to cut down on column maintenance problems. Those extra steps taken to maintain the health and performance of your column will pay dividends in production efficiency and analysis reproducibility. For further reading on the care, maintenance, and troubleshooting of capillary GC columns, consult reference 3.
Simon Jones and Mark Sinnott are GC applications chemists and Allen Vickers is a senior applications chemist with Agilent Technologies at the GC manufacturing site in Folsom, California, USA.
"Column Watch" editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, Delaware, USA and is a member of the Editorial Advisory Board of LCGC Europe.
Direct correspondence about this column to LCGC Europe, Advanstar House, Park West, Sealand Road, Chester CH1 4RN, UK, e-mail: dhills@advanstar.com
References
1. E.J. Guthrie and J.J. Harland, LCGC N. Am., 13(6), 446–455 (1995).
2. R.E. Majors, LCGC N. Am., 16(11), 982–991 (1998).
3. D. Rood, A Practical Guide to the Care, Maintenance, and Troubleshooting of Capillary Gas Chromatographic Systems (Huthig Buch Verlag GmbH, Heidelberg, 1991).





















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

