|Articles|November 1, 2018
- LCGC Europe-11-01-2018
- Volume 31
- Issue 11
Orthogonal Separation Techniques to Analyze (Very) Polar Molecules in Water Samples: Assessing SFC and Reversed-Phase LC–HILIC
Author(s)Thomas Letzel, Stefan Bieber
Supercritical fluid chromatography (SFC) and hydrophilic interaction liquid chromatography (HILIC) are suitable for screening very polar trace organic compounds in environmental water samples. The polarity range of separable compounds in SFC is as broad as the polarity range of the serial coupling of reversed‑phase liquid chromatography and HILIC (reversed-phase LC–HILIC). In this article the orthogonality of SFC and reversed-phase LC–HILIC is assessed. It is shown that both techniques are highly orthogonal and complementary. The parallel use of the two techniques offers additional benefits for the compound monitoring of, for example, pharmaceuticals in surface waters, or the identification of unknown compounds in complex samples.
Advertisement
Supercritical fluid chromatography (SFC) and hydrophilic interaction liquid chromatography (HILIC) are suitable for screening very polar trace organic compounds in environmental water samples. The polarity range of separable compounds in SFC is as broad as the polarity range of the serial coupling of reversedâphase liquid chromatography and HILIC (reversed-phase LC–HILIC). In this article the orthogonality of SFC and reversed-phase LC–HILIC is assessed. It is shown that both techniques are highly orthogonal and complementary. The parallel use of the two techniques offers additional benefits for the compound monitoring of, for example, pharmaceuticals in surface waters, or the identification of unknown compounds in complex samples.
The occurrence of trace organic compounds (TOrCs) in waterbodies is a challenge globally (1). A huge number of residues from pharmaceuticals, pesticides, biocides, chemicals of daily use, and other compound classes can enter the aquatic environment and be detected in waterbodies in ng/L to µg/L concentrations (2,3). Many TOrCs possess a specific mode of action, but environmental concentrations are significantly lower than those usually applied for therapeutic uses. However, the effects of TOrCs on human and environmental health can hardly be estimated. When using impaired waterbodies for the abstraction of drinking water, TOrCs, including persistent, mobile, and toxic compounds, can enter drinking water and negatively impact water quality. As a response to potential risks associated with the presence of TOrCs in environmental waterbodies and general health concerns, many countries have started to investigate and implement strategies to reduce or avoid potential risks posed by TOrCs (4). Although these strategies seem to be hardly comparable at first glance, all rely on powerful analytical tools with mass spectrometric (MS) detection and workflows to identify and monitor TOrCs in waterbodies (5). The monitoring of TOrCs is a complex task because many different compound groups are likely to enter waterbodies. With the introduction of the first version of the “watch-list” under the European Water Framework Directive, pharmaceuticals and hormones such as diclofenac, estrone (E1), 17-beta- estradiol (E2), 17-alpha-ethinylestradiol (EE2), and macrolide antibiotics are also added to routine monitoring on an EU-wide basis (6).
Known TOrCs can be monitored and quantified using target-screening workflows (7). Therefore stable-isotope- labelled internal standards are required and added to the sample prior to analysis. For the identification of unknown compounds, different workflows are available. Depending on the information available about a sample, suspect lists of compounds can be created. The lists contain substances, which theoretically can be contained in the sample (suspect screening) (7). These can be all kinds of water-relevant compounds and metabolite and transformation products of chemicals. Information such as sum formula, MS/MS fragmentation spectra, or hydrophobicity can then be used to search MS data for matching features. To ultimately identify a feature, reference standards are required. To identify MS features as compounds (nontarget screening), which is the reverse strategy of suspect screening, accurate masses, retention times (correlated to hydrophobicity), and other instrumental data are used to collect information about a compound. Ideally this results in one single chemical structure, which can be obtained as a reference standard for final verification.
To detect and identify TOrCs in water samples, reversed-phase liquid chromatography (LC) coupled to MS is commonly used (8–10). This technique is well established, robust, and suitable for the separation and detection of nonpolar to polar analytes (Figure 1). To express polarity, the logarithmic octanol–water distribution coefficient (log D) at pH 7 can be used. With increasing knowledge about the properties and sources of TOrCs, it has become obvious that even more polar compounds than those so far detectable can enter waterbodies. To allow the detection of these compounds, the polarity range of separation techniques is constantly extended. An important aspect is the introduction of polar modifications in stationary reversed-phase LC phases. These allow the range of separable compounds to be extended from nonpolar compounds (log D [pH 7] > 2) towards polar compounds (-2.5 < log D [pH 7] < + 2) (11). However, the separation of very polar analytes (log D [pH 7] < -2.5) is not possible with such stationary phases. Hydrophilic interaction liquid chromatography (HILIC) is well suited for the separation for polar and very polar compounds, but not for nonpolar.
An ideal separation technique should allow compounds in the highest polarity range to be separated within a single run. Since such separations are hardly achievable in LC using just one stationary phase, the coupling of different stationary phases is a viable option. Apart from using two-dimensional chromatography, realized with valves between the two dimensions, serial couplings of two stationary phases can be used too. This simple type of coupling different stationary phase chemistries requires the compatibility of mobile phases used in both separations. Such a serial coupling can be established, for example with reversed-phase LC and HILIC stationary phases, using the same components as mobile phase, but in opposite ratios. This combination is extremely robust and provides highly reproducible separations (12). It allows a reversed-phase LC and a HILIC separation to be combined by using just a T-piece and a second binary pump. This pump is required to adjust mobile phase conditions for the second stationary phase in the coupling and to separately elute compounds from the reversed-phase LC and HILIC analysis. Consequently, reversed-phase LC–HILIC provides the opportunity to separate the full polarity spectrum of analytes from nonpolar to very polar in a single run. A combination of reversed-phase LC–HILIC with mass spectrometric detection is achievable and the positioning of the reversed-phase LC as the first stationary phase allows aqueous samples to be injected directly into the analytical system.
A comparable polarity range of separable compounds can be achieved with supercritical fluid chromatography (SFC) (13). This separation technique typically uses carbon dioxide as the main component of the mobile phase. Because of the very special characteristics of the “green” mobile phase, SFC can provide fast and highly efficient separations.
Material and Methods
An analytical SFC system and a serial coupling of reversed-phase LC and HILIC, both coupled to a time-of-flight MS, were used to separate and detect 274 standard compounds containing pharmaceuticals, industrial chemicals, pesticides, and other environmentally relevant compounds. Analytical conditions are described briefly in the following and can be found in more detail elsewhere (14).
SFC: The SFC system consisted of a binary pump, an auto-sampler, a column oven, a UV detector, and a back pressure regulator (all Agilent Technologies). A 150 mm × 2.0 mm, 5-μm zwitterionic HILIC column (Knauer) was used as the stationary phase in SFC separations. The mobile phase consisted of CO2 and 20-mM ammonium acetate in methanol (as modifier). The flow rate, back pressure, and temperature were held constant at 1.5 mL/min, 150 bar, and 40 °C, respectively. The mobile phase conditions are summarized in Table 1.
Reversed-Phase LC–HILIC: The chromatographic setup of the serial reversed-phase LC–HILIC coupling consisted of two binary pumps, an autosampler, a column oven, and a UV detector (all Agilent Technologies). The first binary pump and the autosampler were connected to a 50.0 mm × 3.0 mm, 2.7-μm nonpolar endcapped Poroshell 120 EC-C18 column (Agilent Technologies). The outlet of this column was connected to a 150 × 2.1 mm, 5-μm, 200 Å ZIC-HILIC column (Merck) and the second binary pump via a T-piece (Upchurch, IDEX Europe GmbH).
The solvents were used in the following way: Solvent A: 10 mM ammonium acetate in 90:10 (v/v) water–acetonitrile; solvent B: 10 mM ammonium acetate in 10:90 (v/v) water–acetonitrile; solvent C: acetonitrile; solvent D: water. Further mobile phase conditions are summarized in Table 2.
Both chromatographic systems were connected to a time-of-flight mass spectrometer (TOF-MS) (Agilent Technologies). An isocratic pump was connected to the outlets of the chromatographic separations, providing a constant flow of reference solution for MS recalibration.
Results and Discussion
The serial coupling of reversed-phase LC and HILIC as well as SFC have recently been tested, established, and compared for the screening for TOrCs in water samples (14). It was shown that both techniques provide a comparable polarity range of separable compounds. The combination with high-resolution MS detection allows TOrCs to be reliably detected in environmental samples. The spectrum of separable and detectable compounds in aqueous samples not only includes nonpolar or polar compounds, commonly analyzed by reversed-phase LC, but also very polar compounds. So far, these compounds have rarely been detected in environmental samples. This allows for the extended environmental monitoring to very polar (and mostly unknown) TOrCs to gain a more comprehensive view on water quality. In addition, it was shown that matrix effects in electrospray ionization are different in both techniques.
In the study mentioned above, which investigated the applicability of reversed-phase LC–HILIC and SFC for the monitoring of TOrCs in water samples, more than 200 compounds were tested in both techniques. The polarity of these analytes ranged from log D (pH 7) = −7.71 to +7.67 (14). Among the separated compounds were pesticides, biocides, industrial chemicals, and other environmentally relevant compounds, such as pharmaceuticals. The separation mechanisms of both techniques are not comparable, resulting in different retention behaviour (see examples in Figure 2). The chromatographic setup of the reversed-phase LC–HILIC coupling allows compounds retained by reversed-phase LC to be differentiated from those retained by HILIC. HILIC-retained compounds elute in the first 15 min, followed by reversed-phase LC-retained analytes. Reversed-phase LC is suitable for the retention of nonpolar and polar compounds (with positive log D values), while all compounds eluted from HILIC possess a negative log D value in this setup. As a result of the serial coupling, polar and very polar compounds like acamprosate, which are commonly not retarded by reversed-phase LC, can be retarded and separated by reversed-phase LC–HILIC. In SFC, no correlation of compound polarity and retention behaviour can be observed. Carbamazepine, the most hydrophobic compound of the selection in Figure 2, elutes first, but close to piracetam, a polar compound. The retention mechanisms might be more complex than those known from LC separations, but SFC separation allows the same polarity range of compounds to be separated as the reversed-phase LC–HILIC coupling by using only one stationary phase.
The orthogonality of LC and SFC is well known and already used (15). It is obvious that reversed-phase LC–HILIC and SFC are orthogonal when plotting the retention times from both applied separations against each other (Figure 3). A linear correlation of retention times in reversed-phase LC–HILIC and SFC could not be verified (r ï½ -0.37), indicating statistical independency. However, both separations complement each other. Compounds with low retention in reversed-phase LC–HILIC (that is, a retention time in HILIC lower than 8 min) are strongly retained in SFC and compounds with low retention in SFC show high retention in reversed-phase LC–HILIC (up to 28 min in reversed-phase LC–HILIC). Interestingly, some very polar compounds show high retention in both techniques (up to 25 min in reversed-phase LC–HILIC and 18 min in SFC). Furthermore, the retention behaviour of compounds eluting until 18 min in reversed-phase LC–HILIC and SFC is remarkable, because the stationary phase material used for both separations was identical, that is, a zwitterionic HILIC material.
Conclusions
As a result, reversed-phase LC–HILIC and SFC can be applied as complementary and highly orthogonal separation techniques. The polarity range of separable compounds is significantly higher than the polarity range accessible by reversed-phase LC separations. This allows the spectrum of detectable TOrCs in water samples to be broadened. For suspects and nontarget screening, the use of both techniques in parallel can contribute strongly to the improvement of data quality and allow the cross-validation of results (7,16). This can be extremely helpful for the differentiation of isomeric compounds, or if limited or no structural information from tandem-MS is available.
Applications that aim to separate and detect a huge number of analytes in a broad polarity range are required in many analytical fields, such as metabolomics, pharmacology, and doping analysis. As a consequence, there is great potential for reversed-phase LC–HILIC and SFC, helping to improve analytical screening and improving knowledge about complex samples.
Acknowledgements
The authors thank Agilent Technologies for the loan of analytical SFC system and Knauer Wissenschaftliche Geräte for the stationary phase used in SFC experiments. Particular thanks to Prof. Dr.-Ing. Jörg E. Drewes, Dr. Giorgia Greco, and Sylvia Große for their contributions and support. The authors acknowledge the financial support from the German Federal Ministry of Education and Research in funding FOR-IDENT (02WRS1456A).
References
- E. Malaj, P.C. von der Ohe, M. Grote, R. Kühne, C.P. Mondy, P. Usseglio-Polatera, W. Brack, and R.B. Schäfer, Proc. Natl. Acad. Sci. U.S.A. 111, 9549–9554 (2014). DOI:10.1073/pnas.1321082111.
- S.D. Richardson and T. Ternes, Anal. Chem.86, 2813–2848 (2014). DOI:10.1021/ac500508t.
- R.P. Schwarzenbach, B.I. Escher, K. Fenner, T.B. Hofstetter, C.A. Johnson, U. von Gunten, and B. Wehrli, Science313, 1072–1077 (2006). DOI:10.1126/science.1127291.
- S. Bieber, S.A. Snyder, S. Dagnino, T. Rauch-Williams, and J.E. Drewes, Chemosphere195, 410–426 (2018). DOI:10.1016/j.chemosphere.2017.12.100.
- S. Bieber, T. Rauch-Williams, and J.E. Drewes, in Assess. Transform. Prod. Chem. by Non-Target Suspect Screen. − Strateg. Work. Vol. 1, J.E. Drewes and T. Letzel, Eds. (American Chemical Society, Washington, DC, USA, 2016) pp. 11–22. DOI: 10.1021/bk-2016-1241.ch002.
- European Commission, Commission Implementing Decision (EU) 2015/495 of 20 March 2015 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council, Official Journal of the European Union, L78/40, 1-3, (2015).
- T. Letzel, A. Bayer, W. Schulz, A. Heermann, T. Lucke, G. Greco, S. Grosse, W. Schüssler, M. Sengl, and M. Letzel, Chemosphere137, 198–206 (2015). DOI:10.1016/j.chemosphere.2015.06.083.
- B.J. Vanderford and R. Pearson, Anal. Chem.75, 6265–6274 (2003). DOI:10.1021/ac034210g.
- N.M. Vieno, T. Tuhkanen, and L. Kronberg, Environ. Sci. Technol.39, 8220–8226 (2005).
- A.A. Deeb, S. Stephan, O.J. Schmitz, and T.C. Schmidt, Sci. Total Environ.601–602, 1247–1253 (2017). DOI:10.1016/j.scitotenv.2017.05.271.
- A. Periat, A. Grand-Guillaume Perrenoud, and D. Guillarme, J. Sep. Sci.36, 3141–3151 (2013). DOI:10.1002/jssc.201300567.
- G. Greco, S. Grosse, and T. Letzel, J. Sep. Sci. 36, 1379–1388 (2013). DOI:10.1002/jssc.201200920.
- S. Bieber and T. Letzel, Gesellschaft Dtsch. Chem. Mitteilungen Der Fachgr. Umweltchemie Und Ökotoxikologie1, 11–16 (2015).
- S. Bieber, G. Greco, S. Grosse, and T. Letzel, Anal. Chem.89, 7907–7914 (2017). DOI:10.1021/acs.analchem.7b00859.
- M.K. Parr, B. Wuest, E. Naegele, J.F. Joseph, M. Wenzel, A.H. Schmidt, M. Stanic, X. de la Torre, and F. Botrè, Anal. Bioanal. Chem.408, 6789–6797 (2016). DOI:10.1007/s00216-016-9805-4.
- S. Bieber, S. Ruppe, S. Grosse, J.E. Drewes, and T. Letzel, in Assess. Transform. Prod. Chem. by Non-Target Suspect Screen. − Strateg. Work. Vol. 1, J.E. Drewes and T. Letzel, Eds. (American Chemical Society, Washington, DC, USA, 2016) pp. 103–117. DOI: 10.1021/bk-2016-1241.ch007.
Thomas Letzel is an analytical chemist with almost 20 years of professional experience in the field of analytical screening techniques using liquid and gas phase chromatography with mass spectrometric detection. He is head of the Analytical Research Group at the Chair of Urban Water Systems Engineering at the Technical University of Munich (TUM), Germany. Dr. Letzel is author and co-author of more than 150 journal papers, book contributions, conference proceedings, and four books.
Stefan Bieber was researcher at the Analytical Research Group at the Chair of Urban Water Systems Engineering at the Technical University of Munich (TUM), Germany. He received his Ph.D. in the year 2017 with studies using polarity extended chromatographic separation techniques and water management strategies. Since 2018 he has been director of the startup company‚ Analytical Research Institution for Non-Target Screening (AFIN-TS), continuing research and giving analytical support for companies in nontarget screening.
Articles in this issue
about 7 years ago
Vol 31 No 11 LCGC Europe November 2018 Regular Issue PDFabout 7 years ago
Stationary Phase Selectivity: The Chemistry Behind the Separationabout 7 years ago
Highlights from the HPLC 2018 Symposiumabout 7 years ago
HPLC Column Maintenance: Tips for Extending HPLC Column LifetimeNewsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.
Advertisement
Related Content
Advertisement


Advertisement
Advertisement
Trending on LCGC International
1
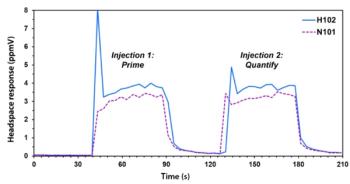
High-throughput Headspace Analysis of Volatile Nitrosamines and their Secondary Amine Precursors
2
Best of the Week: The Scientific Method in the Age of AI, Rewiring the Fundamentals
3
Unlocking Discovery Data: Why a Digital Ecosystem Matters for HT-MS
4
Detecting Estrogen Signatures in MCN Tumors via Targeted LC-MS/MS
5
