Non-aqueous Ion-Exchange Chromatography Using High Acid-base Concentration: A Strategy for Purifying Non-crystalline Pharmaceutical Intermediates
High-quality active pharmaceutical ingredients (APIs) and intermediates are typically delivered using crystallization, distillation, precipitation, and chromatography. However, when organic synthesis delivers a material that will not crystallize, other approaches need to be considered. This article describes a frequently overlooked technique, ion-exchange chromatography (IEX), using non-aqueous mobile phases to purify a non-crystalline intermediate after a reductive amination with D-xylose. IEX delivers a step change in strength and purity of the intermediate allowing successful downstream processing.
- In this project, the intermediate product could not initially be isolated using crystallization, telescoping into the next stage was also unsuccessful and in post-reaction work-up the intermediate product was of low quality.
- A solution involving IEX with a strong cation exchanger offered a means to increase the concentration and purity of the intermediate product which could then be directly used in the next stage of the synthesis.
- IEX offers an alternative to traditional isolation methods.
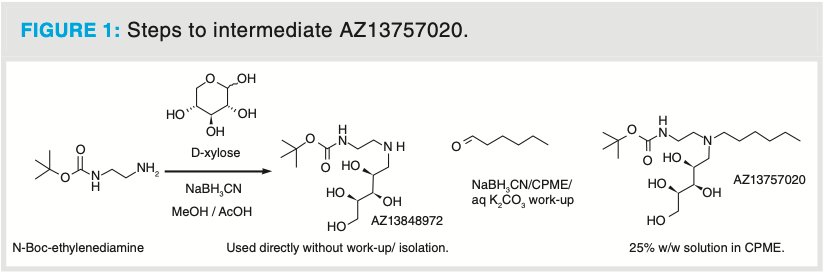
The methods of choice to isolate compounds post-reaction work-up are typically crystallization or precipitation for solids, and distillation for liquids. If these techniques fail to isolate the desired compound at the correct quality, chromatography can be used to improve the purity (1). However, not all compounds are suitable for purification using traditional chromatography. The intermediate AZ13757020 (Figure 1), which is two steps removed from the active pharmaceutical ingredient (API), was synthesized as part of a planned manufacture to deliver 10 kg of the API to support clinical trials within the therapeutic area of cystic fibrosis. However, this intermediate could not be crystallized using classical techniques, leading to delivery of the material with low purity and low quality (% w/w assay). The product is also highly soluble in water, making extraction into an organic solvent difficult. The continuous process development of the intermediate AZ13757020 resulted in each feed (sample) being supplied with varying compositions of the product and impurities. The challenges with a traditional chromatographic purification of this compound were the separation at multi-kilogram scale of a weakly-UV-active product, impurities, and starting materials. We chose to investigate the use of an ion-exchange chromatography (IEX) method using modified silica and high acid-base concentrations in a non-aqueous environment, which also posed a risk of stationary phase instability under such conditions.


Purification Strategy
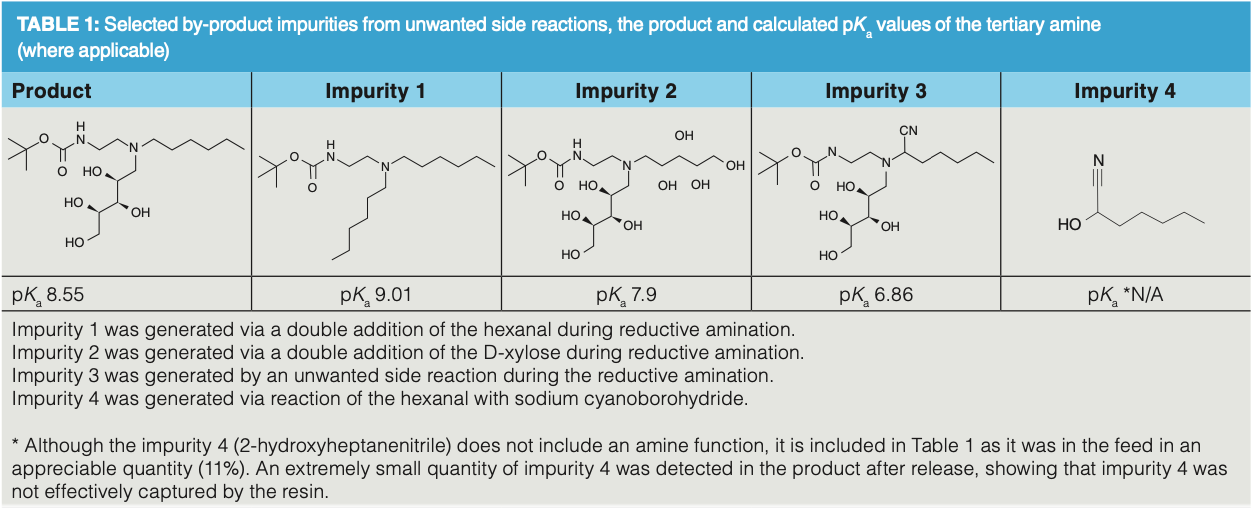
As the product is highly soluble in water, extraction into an organic solvent was difficult; therefore, reversed-phase (RP) chromatography was not a viable option, because lyophilization of multigram fractions was neither practical nor efficient. However, there is a tertiary amine (basic) for IEX and a secondary amide (non-basic) which is not suitable for IEX in the product (Figure 1). This suggested the opportunity to use an acidic ion-exchange resin (strong cation exchanger) to purify the product, exploiting the charge differences between the product and impurities (2). It was anticipated that this could be achieved using a classical load, retain, wash, release process. From the calculated pKa values for the tertiary amines in Table 1, it can be clearly seen that the impurities 1 and 2, and the product would preferentially be bound to a strongly acidic cation exchange resin here because impurities 3 and 4 should be less retentive. Propylsulfonic acid-silica is a strong cation exchanger with minimal non-polar character, used for the retention of basic compounds. It has been successfully used for the “catch and release” purification of amines. When a feed solution containing an amine is passed through a column containing propylsulfonic acid-silica, the amine is retained or “caught” by this column. Non-cationic (non-basic) impurities are not retained and are further removed by washing the column with an organic solvent, such as methanol, acetonitrile, or ethers. The product is subsequently “released” from the column by elution with a solution of ammonia in methanol (methanolic ammonia). Amine salts of weak conjugate acids (such as acetate, and trifluoroacetate) are also exchanged onto the silica-based resin and are released as the free amine during the ammonia/methanol wash. The amine product is then typically isolated by removal of the volatile methanolic ammonia solution by evaporation. The resin was supplied in H+ form, the physical and chemical properties are listed in Table A in Supplementary Information at https://bit.ly/3sKlJh5.

Chemicals: See Table B in Supplementary Information at https://bit.ly/3sKlJh5.
Instrumentation
Milligram scale: A cartridge containing the chosen strong cation exchange was used.
Multi-gram scale: A 25-mm diameter glass column with a maximum bed length of 220 mm (YMC Europe Gmbh, Germany) containing the strong cation exchange resin was connected to a preparative chromatography system (Kronlab GmbH). The system comprised of an AP-250-250-3 quaternary pump, FC-250/250 fraction collector (Armen) with a UV diode array KN-A2700 detection flow cell (Kronlab GmbH). The data capture software, the valve, and flow control operating system was PrepCon 5.0 (SCPA GmbH).
Multi-kilogram scale: A 300-mm diameter glass column with a maximum bed length of 490 mm (YMC Europe), containing the strong cation exchange resin, was connected to a WZ-76200-00 pump (Cole-Parmer). This pump was connected to a custom-built 316 stainless steel multi-port manual solvent manifold. The mobile phase reservoirs were under manual control, when the desired volume of a given mobile phase had been delivered to the column, the next mobile phase was selected as required. Inductively coupled plasma–mass spectrometry ICP-MS: An X-series II ICP-MS system (Thermo Fisher Scientific) was used for ICP-MS analysis. Standards were TraceCert (Sigma-Aldrich). Analysis was run using an external standard method: standards were prepared at 0 (blank), 0.01, 0.1, 1 mg/L (equivalent to 0, 1, 10, 100 ppm in the sample). Samples were prepared at ~10 mg/mL. All standards and samples were prepared in 20% nitric acid with 1 mg/L Yttrium internal standard.
Nuclear Magnetic Resonance (NMR): An A500mHz NMR system (Bruker) was used for NMR analysis. Solvent D-methanol with maleic acid internal standard (20 mg of each in 1 mL).
High Performance Liquid Chromatography (HPLC): The 1100 HPLC system used with associated degasser, quaternary pump, autosampler, column compartment, variable wavelength detector was from Agilent.
Data Handling: Data was processed using Atlas 8.2.3 software (Thermo Fisher Scientific).
Column Packing Method
Milligram scale: The strong cation exchange resin was supplied as
10 g/15 mL pre-packed cartridges. The cartridge was flushed with three column volumes (CVs) of methanol to equilibrate the cation exchange resin.
Multi-gram scale: Approximately 60 g of the strong cation exchange resin was dry packed into a 25-mm glass column. The piston head was then inserted and lowered to 1 cm above the top of the packing. The column was then connected to the UV diode-array detection flow cell. The column was then flushed with three CVs of methanol in backflush mode to equilibrate the column. Backflush mode was used to avoid leaving non-wetted aggregated areas that could encourage the egress of air bubbles. Following the equilibration, the piston head was then lowered until it was just touching the resin bed. This enabled the resin bed length to be measured (190 mm) so a solvated bed density could be calculated. The piston head was then returned to its original height (1 cm above the resin). The solvated bed density was approximately 0.64 g/mL. No swelling of the resin was observed.
Multi-kilogram scale: Approximately 21 kg of the strong cation exchange resin was dry packed into a 300-mm glass column. The piston head was then inserted and lowered to 1 cm above the top of the packing. The column was then connected to the pump and manifold as described in Figure 2. The column was then flushed with three CVs of methanol in backflush mode to equilibrate the column. Backflush mode was used to help avoid leaving non-wetted aggregated areas and help the egress of air bubbles. Upon completion of the equilibration, the piston head was lowered until the resin bed length was 490 mm. Solvated bed density was approximately 0.64 g/mL. No swelling of the resin was observed.
Chromatography (Including Fractionation)
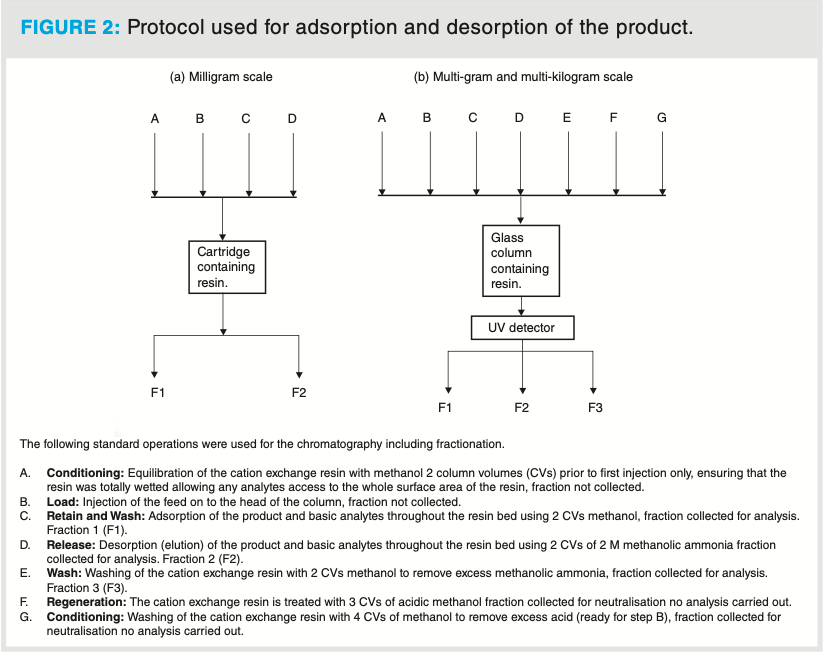
The protocol for adsorption and desorption are shown in Figure 2. Milligram scale: Operations A–D were carried out at the milligram scale (no regeneration, the cartridges were discarded after use). The feed solution (2 mL, approximately 25% w/w solution in cyclopentyl methyl ether [CPME]) was charged to the cartridge in the manner described in Figure 2. The individual fractions were analyzed for the presence or absence of the product on a reversed-phase HPLC (see Supplementary Information at https://bit.ly/3sKlJh5). The fractionation volumes were in the range of 40–60 mL. The product fractions were isolated by reduced vacuum distillation for downstream processing, the quality was confirmed by 1H NMR (see Supplementary Information at https://bit.ly/3sKlJh5).
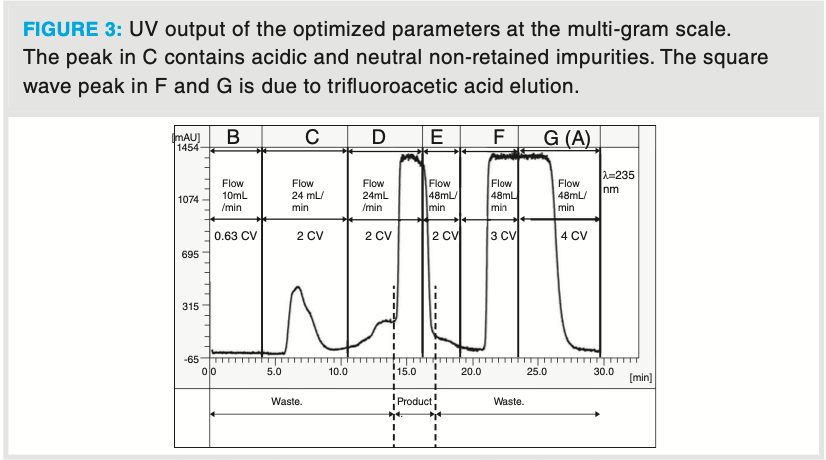
Multi-gram scale: Operations A–G, as described in Figure 2, were performed at the multi-gram scale. These experiments were performed in a continuous manner by monitoring the UV signal and then manually collecting each fraction at the chromatographer’s discretion. Figure 3 shows a typical optimized run, with the unit operations A–G and the conditioning (A/G) steps, load (B), retain and wash (C), release (D), wash (E), regeneration (F) steps. UV output of the optimized parameters at the multi-gram scale. The peak in C contains acidic and neutral non-retained impurities. The square wave peak in F and G is due to trifluoroacetic acid elution. The individual fractions were analyzed to confirm the presence or absence of the product on a reversed-phase HPLC (see Supplementary Information at https://bit. ly/3sKlJh5). The fractionation volumes were in the range of 70–80 mL. The product fractions were isolated by reduced vacuum distillation for downstream processing, and the quality was confirmed by 1H NMR (see Supplementary Information at https://bit.ly/3sKlJh5).
Multi-kilogram scale: Operations A–G, as described in Figure 2, were performed at the multi-kilogram scale. The feed was provided in two separate batches. As a result of slight variations in the batches, a user trial was completed for each batch at the multi-gram scale to determine the optimum loading. Batch 1 was sub-divided into six aliquots and batch 2 into nine aliquots. The separation was performed in a continuous manner with fraction collection at the chromatographer’s discretion. Both batches were successfully processed. The individual fractions were analyzed for the presence or absence of the product using reversed-phase HPLC (see Supplementary Information at https:// bit.ly/3sKlJh5). The fractionation volumes were in the range of 22–35 L. The product fractions were isolated by reduced vacuum distillation for downstream processing, and the quality was confirmed by 1H NMR (see Supplementary Information at https://bit.ly/3sKlJh5).


Results and Discussion
Milligram scale: Initial method development work was completed on a milligram scale, trying to understand and define the load, retain, wash, and release parameters. The desorption (elution) of the product from the resin at this scale initially used 7 M methanolic ammonia. The ammonia concentration was investigated and successfully reduced to 2 M with no impact on the release of the product from the resin or the quality of the material. This change was introduced to reduce the environmental impact of the emissions of ammonia during the vacuum distillation at the multi-kilogram scale.
Concerns were raised whether the silica-based ion-exchange resin would be stable with prolonged exposure to high pH at the proposed larger multi-kilogram scale. Exposure of silica to high pH is known to degrade (hydrolyse) silica-based stationary phases (3). Hence, after successfully demonstrating the process at the milligram scale using the cation exchange resin as described in Figure 2, a potentially more robust polymeric resin, was investigated. It was proposed that a propylsulfonic acid modified styrene-divinylbenzene support would be chemically inert to the prolonged exposure to high and low pH at the larger multi-kilogram scale (4). However, this resin failed to retain the product. Accordingly it was decided to return to the original cation exchange resin chosen, with the same feed source and volume as loaded on to the styrene-divinylbenzene resin was successful.
At this time, it was thought that the resin would be single use, that is, at the multi-kilogram scale, a whole batch (4–5 kg) could be processed in a single run. Therefore, it was decided to continue to the multi-gram scale with the original strong cation exchange resin only on the basis that stability of the resin was not critical.
Feed loads greater than 0.05 g/g of resin (volumes above 2 mL load of the initial sample provided) exceeded the binding capacity of the resin and resulted in breakthrough of the product. (See Supplementary Information at https://bit.ly/3sKlJh5).
Multi-gram scale: Following identification of an outline process at the milligram scale, the focus of the work moved to identification of suitable conditions on a multi-gram scale.
Initial experiments had varying degrees of success because the product was not consistently retained. It was observed that as an injection “plug” passed through the column, the feed in CPME diluent, remained in a tight band throughout elution, indicating that CPME had minimal miscibility with the eluent, methanol (MeOH). This raised concerns that the large difference in polarity between CPME and methanol may have affected the efficiency of the ion-exchange process. As part of the investigative work it was found that when the CPME feed solution was left standing for several hours, the feed was clearly biphasic. A non-homogenous feed would have different concentrations of the product in either layer, potentially leading to varying quantities being loaded onto the column for each injection. This could have been one of the contributing factors to the inconsistent retention of the product. Moving from CPME to MeOH (as feed diluent) produced a homogenous feed, eradicating the biphasic feed issue, and it was thought that this would lead to more consistent loadings and chromatography. This conjecture was later proved to be unfounded; the reason why many of the experiments failed was not due to the biphasic feed.
It was unknown if the resin would be stable to the high pH (2 M methanolic ammonia, product release (step D in Figure 2) or if the resin could be acid regenerated. Basic or acidic modifiers are usually present in the order of 0.1/0.5% v/v in chromatography eluents (5), but in this case the ammonia was present at 3.5% (2 M methanolic ammonia pH≈10.5). For resin regeneration, an acid soluble in methanol was required; water was not an option as the product is highly soluble in water. Hydrochloric acid could not be used as it was not compatible with the equipment at this scale and at the multi-kilogram scale (6). Sulphuric acid was not explored as it is a diprotic acid. Methanesulphonic acid resulted in the precipitation of ammonium methanesulphonate, which is insoluble in methanol and hence would have blocked the preparative system tubing. It was proposed that 2 M acetic acid in methanol would be sufficiently acidic for regeneration without corroding the 316-stainless steel tubing, steps (E–G) were carried out. Experimental work showed that the resin could not be regenerated using 2 M acetic acid. The resin was then subjected to 2 M TFA (23% TFA in MeOH) regeneration, steps (E–G). This successfully regenerated the ion-exchange stationary phase, however, the chromatography remained inconsistent.
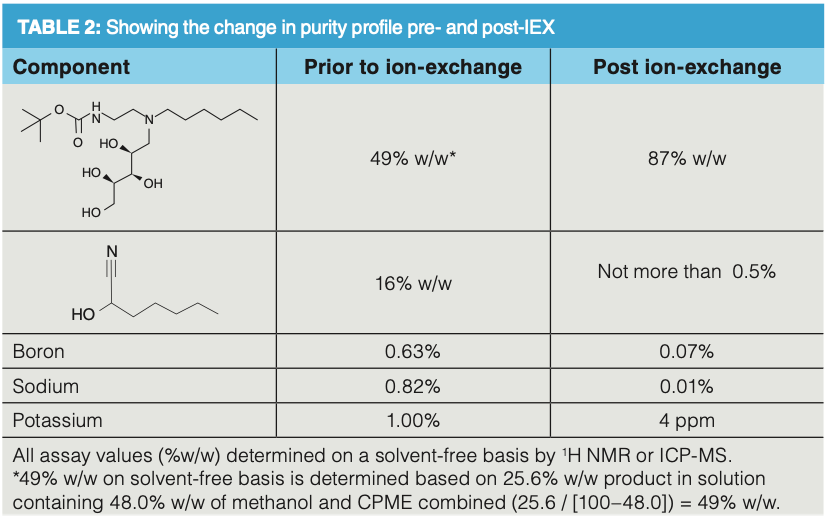
As this is a cationic exchange process it was postulated that the inconsistent chromatography could be attributed to the presence of other cationic species present in the feed, for example, sodium and potassium, as overloading had been ruled out. This led to a sample of the feed and a sample from a successful run being analyzed by ICP (see Supplementary Information at https://bit.ly/3sKlJh5). The results clearly showed that the feed contained large quantities of cations (pre-IEX), and these were significantly reduced by the resin in the successful run (post-IEX). These results indicated that the failure of many of the experiments was a result of the presence of competing, smaller cationic impurities (non-amines). These stronger binding, small positively charged cations would preferentially bind with sulphonic acid ion-exchange sites, and when all the small cationic impurities were bound to the ion-exchange stationary phase, the amine-product would then bind to the remaining sites.
When these ICP results became available, the multi-kilogram manufacture of the product was already in progress and there was insufficient time or resource to develop a process to remove or reduce the cations present. Therefore, it was decided to perform a user trial of each batch at the multi-gram scale, to find a loading that would successfully scale up to the multi-kilogram scale. The user trial of each batch was based on trialling a volume at the multi-gram scale to find the optimum loading for the multi-kilogram scale. Using this data, the number of aliquots required to process a batch was calculated (see Supplementary Information at https://bit.ly/3sKlJh5).
The results from the user trials demonstrated that the CV approach applied at multi-gram scale could be used to define the optimum load volume at multi-kilogram scale, making scale-up straight forward. The CV at the multi-kilogram scale (300 mm i.d. × 490 mm bed length) was calculated at 35 L (πr2h) for the first aliquot of the first batch. This was known to be an overestimation to take into account the dead volume (interstitial volume and pore volume with respect to the resin) of the system, ensuring that no product would be lost and that the regeneration and washing steps were not underestimated. After the first aliquot, it became clear that this calculated volume was excessive, and the actual CV was determined to be 22 L.
The 2 M methanolic ammonia base strength proved strong enough to remove the product and associated organic basic impurities (see Table 1). Serendipitously, the resin bound potassium, boron, and sodium were not displaced, otherwise these cations would have posed an issue for the downstream processing.

The most useful UV wavelength to monitor the chromatography was determined to be 235 nm. This ensured that the elution of the product could be observed without interference from the methanolic ammonia mobile phase/wash solvent (ammonia has a UV cutoff at 217 nm).
Multi-kilogram scale: During steps A–G (Figure 2) of the first run at this scale, the column temperature was monitored for excess heat because it was believed that the regeneration step would have the largest heat generation (acid-base reaction). However, this was found not to be the case, with the release step D, generating so much heat that the metal fittings on the exit line became quite warm. A reduction in flow rate controlled this heat generation.
An aliquot of feed was processed in 4 h. Hence, the processing of 17 aliquots took 68 h. The throughput was 284 mL of feed/kg resin/day (60 g of product/kg resin/day).
The pump flow rates were set between 1.0–1.5 L/min, which was not a linear scale up of the flow rates from multi-gram to multi-kilogram scale. The flow rates were governed by the maximum back pressure that the glass column could withstand (the maximum safe working pressure supplied by the manufacture was 5 Barg). For the aliquot sub-division of each batch, see Supplementary Information at https://bit.ly/3sKlJh5.
Conclusion
The use of IEX in 100% organic mode for scale-up to multi-gram has been successful to deliver a step change in the quality of a non-crystalline intermediate that cannot be purified by standard methods. This has been successfully applied to AZ13757020 converting material of 49% w/w to 87% w/w. This allowed the high-quality material to be successfully used in subsequent manufacturing stages. As demonstrated in this article IEX, is a powerful, productive, and effective technique and should be considered as part of the toolbox in process development and manufacturing for purification of pharmaceutical materials.
Acknowledgements
The authors are extremely grateful to the following colleagues: Jeremy Parker (for his valuable advice, guidance, and support), Malvika Sardana, and Neil Sumner for their help with the preparation of this manuscript. Thanks also to Peter Moore and Joanna Hemming Taylor.
References
- A.M. Katti, E.V. Dose, and G. Guiochon, J. Chromatogr. A. 540, 1–20 (1991).
- J.S. Fritz, Anal. Chem. 59(4), 335A–334A (1987).
- J.J. Kirkland, M.A. van Straten, and H.A. Claessens, J. Chromatogr. A. 691(1–2), 3–19 (1995).
- M.V. Srikanth, S.A. Sunil, N.S. Rao, M.U. Uhumwangho, and K.V. Ramana Murthy, J. Sci. Res. 2(3), 597 (2010).
- M.C. García, J. Chromatogr. B. 825(2), 111–123 (2005).
- British Stainless-Steel Association https:// www.bssa.org.uk/topics.php?article=30
AUTHORS
Andrew Ikin is associate principal scientist at AstraZeneca and has delivered APIs and intermediates from the nano-gram to the metric tonne. He has over 29 years’ experience in the pharmaceutical industry, and has specialized in large-scale chromatography for 16 years, both in batch mode and simulated moving bed (SMB). He has developed in-house separations (normal phase and RP) that have delivered separations from the gram to 100 kg, supporting clinical trials for toxicity studies, right through to Phase III clinical trials within a strict good manufacturing practice (GMP) envelope.
Lee Timms is a senior large-scale laboratory chemist at AstraZeneca, with 20 years of experience running multi-step development manufacturing campaigns, with an interest in flow chemistry at scale and the use of process analytical technology (PAT).
Andrew Stark graduated from Strathclyde University, Scotland, UK, and has been a process chemist with AstraZeneca for 19 years. He has worked on dozens of projects from mg- to multi-Kg scale, and understands the importance of having access to chromatography during the early phase of drug substance development.
Craig Stewart has been a synthetic process development chemist since 2002, within AstraZeneca’s Pharmaceutical Sciences department in Macclesfield, UK.
Direct correspondence to: amatheson@mjhlifesciences.com


 for the Characterization of Therapeutic Monoclonal Antibodies and Related Products, Part 1: Theoretical Aspects")

















