Multi-Toxin Determination in Food – The Power of "Dilute and Shoot" Approaches in LC–MS–MS
LCGC Europe
Multi-Toxin Determination in Food – The Power of "Dilute and Shoot" Approaches in LC–MS–MS
A liquid chromatography tandem mass spectrometry (LC–MS–MS) “dilute and shoot” method for the determination of 331 (toxic) secondary metabolites of fungi and bacteria has recently been optimized and validated for different food matrices (1) according to the guidelines established in the Directorate General for Health and Consumer Affairs of the European Commission (SANCO) document No. 12495/2011. This article will provide useful tips for smooth validation of multi-analyte LC–MS–MS methods and summarizes important validation outcomes for 295 analytes, including over 200 mycotoxins. The second part of this article focuses on the performance of the method in proficiency testing with an emphasis on difficult matrices. Z-scores obtained with this method were between -2 and 2 in 368 out of 408 cases for maize, wheat, triticale, bran, nuts, baby food, raisins, figs, coffee, liquorice, and hot pepper. From these results it can be concluded that quantitative determination of mycotoxins by LC–MS–MS based on a “dilute and shoot” approach is also feasible in the case of complex matrices such as such as pepper, coffee, or liquorice.


Photo Credit: Dr. Roman Labuda
Spoilage of food and feed is still a global problem, leading to enormous annual losses of several hundred million tonnes. In relation to that, microscopic filamentous fungi are considered as one of the most feared agents because of mycotoxin production (2). Mycotoxins are low-molecular-weight, secondary metabolites of mold, and are toxic to animals and humans even in low concentrations (3). The European Union (and many other countries) has therefore established maximum levels for the most important mycotoxins in food (4). Such regulations can be laid down or amended only based on long-term occurrence monitoring and availability of toxicological data followed by exposure risk assessment. The European Food Safety Authority (EFSA) organizes a continuous call for data collection about the occurrence of already regulated (aflatoxins, ochratoxin A, deoxynivalenol, HT-2 toxin, and T-2 toxin) and so-far unregulated mycotoxins (nivalenol and ergot alkaloids) (5). Further collection of accurate data is also needed for enniatins, beauvericin, sterigmatocystin, Alternaria toxins, citrinin, and phomopsins for human and animal exposure assessment (6).
The increasing requirements for accurate and reliable occurrence data for more than one toxin has led to great progress in the development of highly selective, sensitive, and accurate LC–MS–MS methods for multi-mycotoxin determination in food and feed (7). A range of methods has been published on the identification and accurate quantification of single or chemically related mycotoxins in several matrices (8). However, the number of validated multi-analyte methods covering mycotoxins of different classes is limited for several reasons, beginning with sample preparation. The extraction solvent used has to solve a broad range of chemically unrelated toxins, and sophisticated cleanups have to be omitted to avoid discrimination of some compounds. Unspecific extraction and the reduction of sample cleanup to a minimum can result in suppression or enhancement of analyte responses during the ionization process. So-called matrix effects are a major challenge in successful development of reliable, quantitative multi-analyte methods. Therefore, considerable efforts to control matrix effects should be performed to obtain accurate results. However, common approaches for counteracting matrix effects (unspecific QuEChERS clean-up, standard addition, matrix-matched standard addition, SIDA [stable isotope dilution assay]) are not feasible for LC–MS–MS methods covering more than 200 mycotoxins. In this case, injection of diluted extracts on a highly sensitive LC–MS–MS instrument has been shown to be the most effective approach (1).
Performing in-house validation is necessary for reliable quantification at a high level of trueness, preferably according to international guidelines. However, validation of each matrix that should be analyzed for around 300 substances is costly and time-consuming. The SANCO document for development of multi-analyte methods in pesticides residue analysis recommends that at least one representative commodity from each commodity group should be validated and evidence for fitness of purpose should be provided (9). This approach, which was successfully applied in pesticide residues analysis, has also been followed for the validation of the developed multi-mycotoxin LC–MS–MS method (1).
Another difficulty in quality assurance of multiâmycotoxin LC–MS–MS data is the limited or even non-availability of certified reference materials containing multiple structurally unrelated mycotoxins. Therefore, participation in proficiency testing is the only option to prove the long-term performance of the method.
The objective of this article is: (i) to provide useful tips for smooth validation of multi-analyte LC–MS–MS methods; (ii) to summarize the important validation outcomes; and (iii) to present the long-term performance of this “dilute and shoot” method in the proficiency testing with a special focus on “difficult” matrices.
Experimental
Chemicals and Consumables: Methanol, acetonitrile, ammonium acetate (all LC–MS grade quality), and glacial acetic acid (p.a.) were purchased from Sigma Aldrich. A Purelab Ultra system (ELGA LabWater) was used for further purification of reverse osmosis water.
Standards of fungal and bacterial metabolites were obtained either as gifts from various research groups or from the following commercial sources: Romer Labs Inc., Sigma-Aldrich, Iris Biotech GmbH, Axxora Europe, and LGC Promochem GmbH.
Method Validation According to the SANCO Document: Detailed information about method validation is provided in (1). Briefly, four different model matrices were chosen for spiking experiments - apple puree (high water-containing matrix), hazelnuts (high fat content), maize (high starch content), and green pepper (complex or “difficult” matrix) (9). Blank ground samples of 0.50 g were spiked with solutions containing all analytes and left overnight in darkness. Samples were then extracted with 2 mL of acetonitrile/water/acetic acid 79:20:1, v/v/v for 90 min on a rotary shaker (GFL) and subsequently centrifuged for 2 min at 3000 rpm. The extracts were transferred into glass vials using Pasteur pipettes, and 350 μL aliquots were diluted with the same volume of acetonitrile/water/acetic acid 20:79:1, v/v/v. Finally, 5 μL of the diluted extract was injected into a LC–MS–MS system without further pre-treatment. Spiking experiments were performed at four levels in five repetitions of each, resulting in relative concentrations of 1:3:10:30 of the final diluted extracts. External and matrixâmatched calibrations were prepared by dilution of the final working solution with acetonitrile/water/acetic acid (49.5/49.5/1, v/v/v) and diluted blank extracts of each model matrices, respectively, at the levels corresponding to spiked samples. In addition, one calibration point below and one above the spiking concentration range were prepared in anticipation of the expected matrix induced signal suppression and enhancement of target analytes.
The peaks were integrated and linear, 1/x weighted, calibration curves were constructed from the data obtained from the analysis of each sample type (spiked sample, neat solvent standard, spiked extract) using MultiQuant 2.0.2 software (Sciex) to evaluate the linearity of the method. Further data evaluation was performed in Microsoft Excel 2007. All the other performance characteristics of the method (recovery, apparent recovery, repeatability, and matrix effects) were evaluated at each spiking level for all model matrices (1).
Instrumental Parameters: Detection and quantification was performed with a QTrap 5500 MS/MS system (Sciex) equipped with a TurboV electrospray ionization (ESI) source and a 1290 series UHPLC system (Agilent Technologies). Chromatographic separation was performed at 25 °C on a 150 × 4.6 mm, 5-μm Gemini C18-column equipped with a C18 security guard cartridge, 4 × 3 mm i.d. (Phenomenex). Elution was carried out in binary gradient mode. Both mobile phases contained 5 mM ammonium acetate and were composed of methanol/water/acetic acid 10:89:1 (v/v/v; eluent A) and 97:2:1 (v/v/v; eluent B), respectively. After an initial time of 2 min at 100% A, the proportion of B was increased linearly to 50% within 3 min. Further linear increase of B to 100% within 9 min was followed by a hold-time of 4 min at 100% B and 2.5 min column re-equilibration at 100% A. The flow rate was 1000 μL/min. ESI–MS–MS was performed in the scheduled multiple reaction monitoring (sMRM) mode both in positive and negative polarities in two separate chromatographic runs. The sMRM detection window of each analyte was set to the respective retention time ±27 s and ±42 s in positive and in negative mode, respectively. The target scan time was set to 1 s. The settings of the ESI source were as follows: source temperature: 550 °C; curtain gas: 30 psi; ion source gas 1 (sheath gas): 80 psi; ion source gas 2 (drying gas): 80 psi; ion-spray voltage: −4500 V and +5500 V, respectively; collision gas: (nitrogen) medium. Confirmatory identification was obtained through the acquisition of two sMRMs per analyte (with the exception of moniliformin and 3-nitropropionic acid that each â¨exhibit only one fragment ion) (1), which yields 4.0 identification points according to commission decision 2002/657/EC (10).
Method Trueness: The trueness of the method is shown by participation in proficiency tests organized by Bipea (Gennevilliers, France) and by the Institute of Science of Food Production of the National Research Council of Italy (ISPA-CNR, Bari, Italy) (15).
Results and Discussion
Useful Tips for the Experimental Set-up of the Validation Process and Validation Data Evaluation: As no guidelines and recommendations suitable for validation of multiâmycotoxin methods (or multi-analyte methods in general) have been laid down so far, the recommendations provided by SANCO for pesticides have been followed (9). However, slight amendments were necessary to improve validation outcomes, minimize working time for sample preparation, and lower validation costs.
Spiking Concentrations: SANCO suggests validation at two concentration levels using five repetitions, one level close to limit of quantification (LOQ) and one higher. As the final working mixture of analytical standards contained 331 analytes at various concentrations, we decided to validate four levels, each at five repetitions. Such experimental setup amendment prevented a potential non-linearity of the method for some analytes and discrimination of analytes with high LOQs.
Miniaturization: The whole sample preparation procedure can be miniaturized for validation purposes to decrease the amount of standards needed for spiking. For example, we used 0.5 g of blank sample material plus 2 mL of extraction solvent for validation, but in routine analysis and proficiency testing, 5 g of sample was extracted with 20 mL of extraction solvent instead. Be careful: High sugar content matrices (for example, raisins) cannot be miniaturized to 0.5 g because of inhomogeneity (1).
Method Accuracy: This was evaluated based on apparent recoveries instead of extraction recovery because we use neat-solvent calibration and spiked samples in daily routine analysis rather than matrix-matched standards. The reason for this is that it is very difficult to find a blank matrix for all analytes involved in the method. Apparent recovery expresses both extraction efficiency and matrix effects, and therefore it contains more relevant and “real-life” information (1). In routine analysis, data are also corrected for apparent recovery.
Matrix Effects: As SANCO provides no recommendations about the acceptable range for matrix effects, the range of 90–110% was considered to have no effect. Matrix effect evaluation should be performed using two approaches: “traditional slope” and “one calibration point”. “Traditional slope” evaluation is commonly used during validation. The slope obtained from matrixâmatched standards is compared to the one obtained from neat solvent standards. However, evaluation of matrix effects by comparison of each calibration point instead of the slope better reflects the real extent of matrix effect across the whole concentration range. Therefore, we recommend using both approaches during validation (1).
Limit of Quantification (LOQ): Evaluation of LOQ according to SANCO is highly recommended for multi-analyte methods. LOQ is estimated as the lowest spiking level (LL) that allows reliable detection of all five replicates meeting the performance criteria of RSD < 20% and mean recovery of 70–120% at both SRM transitions. LOQs obtained by the LL approach did not differ significantly from LOQs calculated from signal-to-noise in our experience (1).
Validation Summary: Following the guidelines laid down by SANCO for pesticide analysis, convenient validation data of our method were obtained for 295 out of 331 analytes. The remainder of the compounds could not be reliably validated for several reasons: (i) non-availability of analytical standards; (ii) instability of an analyte in the final working solution; and (iii) low concentration of analytical standard for spiking. Validation for some analytes was done only semi-quantitatively; a fungal extract was used for spiking instead of analytical standards that were not commercially available (1).


The distribution of apparent recoveries and matrix effects throughout the set of 295 analytes in apple puree, hazelnuts, maize, and green pepper at the lowest and the highest spiking levels is displayed in Figure 1 and 2, respectively. The highest validation level corresponds to a 1:10 dilution of the final working solution of the analytical standards. As the lowest validated level, the lowest spiking level reliably detectable at all five repetitions (RSD < 20) on both SRM transitions was considered. As expected, green pepper was the most difficult matrix. Only 20% of analytes met acceptable apparent recoveries of 70–120%, as recommended by SANCO (9). Moreover, 25% of the compounds were not detected in green pepper at any level lower than the highest validated level. With regards to the other matrices, more than 50% of compounds were in the acceptable range of 70–120%. For the analytes out of this range, either high matrix suppression or enhancement or â¨low extraction efficiency was observed. More detailed information is provided in (1).

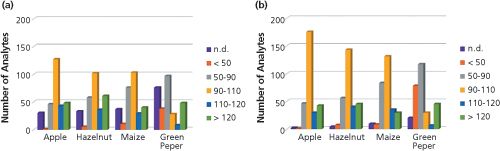
Any influence of matrix effects was observed for all matrices, albeit to a varying extent, which was strongly dependent on the analyte/matrix combination (see Figure 2). Both matrix suppression and enhancement were observed. In general, the lowest matrix effects were obtained in apple puree and the highest in green pepper. Only 10% of analytes were not affected by matrix effects in green pepper.
Method precision was proven by repeatability of five repetitions at the highest and lowest level. Less than 5% of compounds were above the RSD of 20% in apple puree, maize, and hazelnuts. In green pepper, the RSD was not below 20% for 11% of all validated analytes.
Method Trueness - Proficiency Testing: To check for the trueness of the results obtained by our “dilute and shoot” LC–MS–MS approach, we have been participating in a number of laboratory proficiency testing (PT) schemes organized by various institutions (Bipea, CODAâCERVA, FAPAS) since 2010. Regular PT participation enables method drawbacks to be highlighted, helps to detect if something is out of control (for example, spoilage of a standard), and assists in the improvement of the overall method performance. Furthermore, analysis of uncommon and difficult matrices such as raisins or spices adds information about method accuracy in complex or even non-validated matrices. Generally, the spectrum of analytical methods used by participating laboratories varies from ELISA through conventional HPLC coupled to fluorescence detection (FLD) to advanced LC–MS–MS or LC–high resolution mass spectrometry (HRMS)-based methods. Therefore, each successful PT participation has proved that the developed multi-mycotoxin method can reliably compete with other methods in terms of accuracy. A few examples of PTs are discussed below.

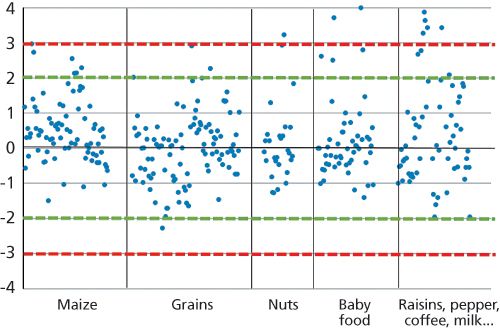
A total of 408 z-scores have been calculated out of the reported data within our continuous PT participation, and 368 values were in the acceptable range of -2 < z < 2. Only 23 z-scores were considered as questionable (between -3 and 3) and 17 were unacceptable (z < -3, z > 3). Figure 3 summarizes the z-scores of our multi-mycotoxin LC–MS–MS method in Bipea PT (11), where multiple mycotoxins were determined in maize, grains, nuts, baby food, and “difficult” matrices. The distribution shows that the majority of outliers (z-scores) were calculated for baby food, which exhibited concentrations very near the LOQ, and difficult matrices.

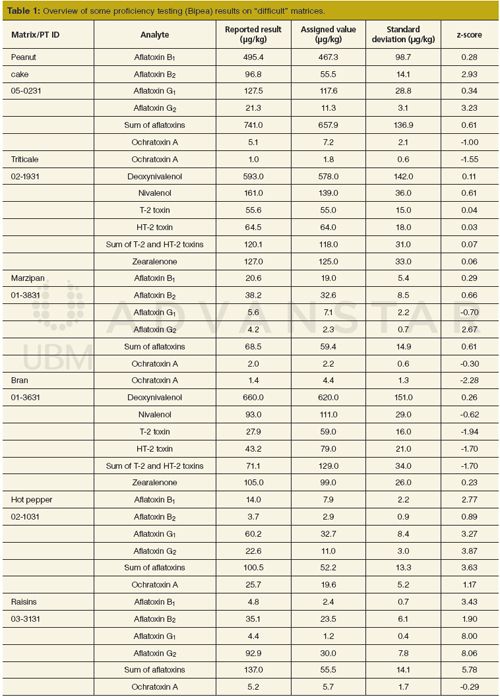

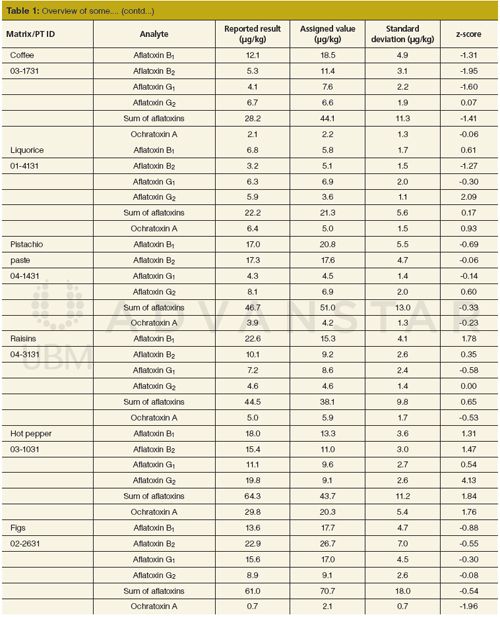
The performance of the method in some “difficult” matrices is presented in Table 1. Although, matrices such as raisins, liquorice, or figs have never been fully validated, most of the obtained results were within the acceptable z-score range (-2 < z < 2). However, the z-scores for aflatoxins in hot pepper (02-1031) were out of the acceptable range. The reported results were evaluated on the external neat-solvent calibration curve and corrected on the apparent recovery obtained from spiking of the validated results of green pepper (12). Another very complex matrix was raisins. The sample of raisins with identification number of 03-3131 was not successfully analyzed for aflatoxins. In this case the results reported by laboratories were dispersed widely. During the homogeneity testing and checks between bags, some differences for aflatoxins G1 were found (13). Obviously, most of the participants did not homogenize the whole obtained batch. The assigned value was not calculated from the data of participating laboratories, but the reference laboratory measurements instead. It should be noted that reported results for aflatoxins in raisins (04-3131) analyzed later were in agreement with other laboratories, yielding acceptable z-scores (14).

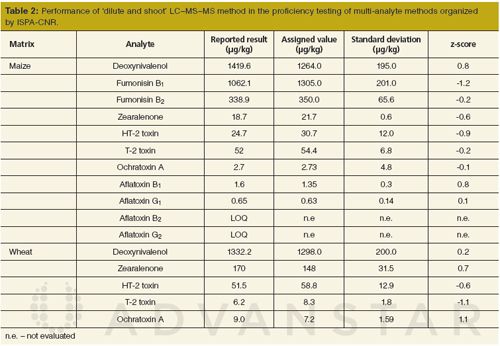
In 2014, ISPA-CNR organized a proficiency test on mycotoxins (15). It was aimed at laboratories willing to test the trueness of their multiâmycotoxin methods. Therefore, mainly LC–MS–MS methods were used for the quantitative determination of deoxynivalenol, zearalenone, HT-2 and T-2 toxins, fumonisins, aflatoxins and ochratoxin A in maize and deoxynivalenol, zearalenone, HT-2 and T-2 toxins, and ochratoxin A in wheat within this proficiency testing scheme. The performance of our method is summarized in Table 2. The developed LC–MS–MS multiâmycotoxin method was successful, yielding z-scores within the range of -2 < z < 2 (15). Concerning aflatoxin B2 and aflatoxin G2 in maize, no statistical evaluation was reported because of a lack of sufficient data.
Conclusions
This article presents information on the background and the personal experiences which have been gained during the validation of a “dilute and shoot” multi-analyte LC–MS–MS method for the determination of 331 (toxic) secondary metabolites of fungi and bacteria in food. In addition, guidance on proper method validation of a multi-analyte LC–MS–MS method is provided.
The performance of our “dilute and shoot” method in proficiency tests has been satisfactory. The routine participation in proficiency testing has proved that the developed and validated LC–MS–MS method is able to provide accurate results for regulated mycotoxins in the case of “difficult” matrices such as raisins, figs, or liquorice.
References
(1) A. Malachová, M. Sulyok, E. Beltrán, F. Berthiller, and R. Krska, J. Chromatogr. A1362, 145–156 (2014).
(2) http://www.mycotoxins.org/node/56 (accessed on 15 July 2015).
(3) J.W. Bennett and M. Klich, Mycotoxins Clin. Microbiol. Rev.16, 497–516 â¨(2003).
(4) Commission regulation (EC) No. 1881/2006 http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:364:0005:0024:EN:PDF (accessed on 15 July 2015).
(5) EFSA. http://www.efsa.europa.eu/en/search.htm?search=call&text=call (accessed on 15 July 2015).
(6) CEN mandates, Call for tender for project leaders for the development of standardized methods for the analysis of mycotoxins in food (available online since 10 May 2013) file:///C:/Users/ataml/Downloads/Call_520_CENwebsite_2013_05_10.pdf
(7) Web of Science, Thomson Reuters.
(8) P. Zöllner and B. Mayer-Helm, â¨J. Chromatogr. A1136, 123–169 â¨(2006).
(9) Document SANCO/12495/2011. http://ec.europa.eu/food/plant/plant protection products/guidance documents/docs/qualcontrol en.pdf (accessed on 10 December 2013).
(10) 2002/657/EC. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF (accessed on 15 July 2015).
(11) Bipea, Provider of Proficiency Testing Programs. http://www.bipea.org/â¨content/pt-programs (accessed on â¨15 July 2015)
(12) Bipea, Interlaboratory comparisons report on hot pepper (02-1031), 31a-133-Mycotoxins-Aflatoxins and Ochratoxins, 12 April 2013.
(13) Bipea, Interlaboratory comparisons report on raisins (03-3131), 31a-134-Mycotoxins-Aflatoxins and Ochratoxins, 24 July 2013.
(14) Bipea, Interlaboratory comparisons report on raisins (04-3131), 31a-140-Mycotoxins-Dried Fruits, Spices and Other Products, 16 October 2014.
(15) A. De Girolamo, B. Ciasca, J. Stroka, S. Bratinova, A. Visconti, and V.M.T. Lattanzio, Report of the 2014 Proficiency Test for LC-MS(MS) multi-mycotoxin methods, June 2015. http://www.ispacnr.it/?p=479
Alexandra Malachova studied analytical chemistry at the Department of Food Analysis and Nutrition, Faculty of Food and Biochemical Technology of the University of Chemistry and Technology (UCT Prague), Czech Republic. She received her PhD degree in 2012. She currently works as a postdoc in the Christian Doppler Laboratory for Mycotoxin Metabolism and the Center for Analytical Chemistry at the Department of Agrobiotechnology (IFA-Tulln) of the University of Natural Resources and Life Sciences, Vienna (BOKU). She is an expert in LC–MS, including high resolution MS. Her main field of expertise is in development, optimization, and validation of LC–MS-based methods for multiple analytes or methods for analysis of complex matrices.
Michael Sulyok is Head of the Laboratory for Multi-Toxin Screening at the Department of Agrobiotechnology (IFA-Tulln) at BOKU. His publication from 2006 on a validated LC–MS–MS based method for the determination of 39 mycotoxins in wheat and maize is the most often cited original paper in the field of mycotoxins from the past 10 years. This method has been continuously expanded to become the world’s most comprehensive approach for the quantitative determination of mycotoxins and fungal metabolites in food, feed, and damp indoor environments. The analytical quality of the method is continuously monitored by participation in proficiency tests and multi-toxin inter-laboratory comparison studies with the aim to prove that the accuracy of the LC–MS–MS-based “dilute and shoot” approach may compete with conventional methods. Dr. Sulyok has (co-)authored more than 100 SCI publications (h-factor 27).
Eduardo Beltran obtained an International PhD degree at University Jaume I (Castellon, Spain) in April 2014. His research was based on the development of analytical methods for the determination of natural toxins in food and environmental samples by liquid chromatography coupled to tandem mass spectrometry with triple quadrupole analyzers. He has published around 15 peer-reviewed articles in international journals and 20 communications in workshops and congresses. He has performed pre-doctoral and post-doctoral research at the Center for Analytical Chemistry-IFA (Tulln, Austria). Current research is mainly focused on the application of LC–MS for the development of analytical methods for quantification and confirmation of different compounds (toxins, hormones, pesticides) in different matrices at very low levels.
Franz Berthiller is an associate professor at the University of Natural Resources and Life Sciences, Vienna (BOKU) and Head of the Christian Doppler Laboratory for Mycotoxin Metabolism. In 2006 he received the Brigitte-Gedek-Award from the German Society of Mycotoxin Research for his PhD thesis on masked mycotoxins. He continued his research in the laboratory of Rudi Krska at the IFA-Tulln, as well as some months abroad at the Danish Technical University and the Food Research Division of Health Canada in Ottawa. As an expert in the areas of (modified) mycotoxins and LC–MS, his scientific output includes about 100 SCI publications.
Rudolf Krska is full professor for (Bio-)Analytics and Organic Trace Analysis and is head of the IFA-Tulln with more than 180 co-workers at the University of Natural Resources and Life Sciences, Vienna (BOKU). From 2009 to 2010 he worked for one year as Chief of Health Canada´s Food Research Division in Ottawa. He has received 10 scientific awards and is (co-) author of more than 250 SCI publications (h-index: 42). His current research interests are in the area of plant-fungi metabolomics and novel mass spectrometric methods for the determination of multiple mycotoxins including their conjugation and transformation products in food, feed, and other biological matrices.













Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.




