Method Robustness and Reproducibility for Monoclonal Antibody Purity Analysis
Intact and subunit analysis by reversed-phase liquid chromatography (RPLC) is a common method for purity assay of monoclonal antibodies (mAbs). Monitoring these data in a quality control environment requires a reliable method to ensure any critical quality attributes are identified during development and production. This article describes the robustness assessment for the analysis of National Institute of Standards and Technology (NIST) mAb, intact and reduced, using a wide pore C4 LC. Method parameters such as temperature, acidic modifier, and gradient slope are assessed to determine their effect on method performance.
Monoclonal antibodies (mAbs) are well‐established therapeutics, with a variety of different analytical methods associated with purity assays. These include many liquid chromatography (LC) and non‐LC related techniques. However, one particularly attractive analytical technique common for mAb purity is reversed‐phase high performance liquid chromatography (RP‐HPLC). This analysis can be performed in a fairly short time, and is relatively high resolution, capable of separation of hydrophobic variants including oxidation, glycoforms, and lysine variants. As such, intact reversed‐phase methods can be implemented early during development, for example, during lead selection and optimization. Coupling RP‐HPLC to high resolution mass spectrometry (HRMS) facilitates in the characterization of these unknown variants, which is beneficial because the method transitions to downstream for stability and quality control lot‐release testing.
Consequently, it behooves the analytical method developer to ensure that the RP‐HPLC method is robust. This is especially true if a method originates from a platform method; that is, one which is applicable to many different analytes. This is most certainly a viable strategy during early development, and one that is encouraged with intact RP‐HPLC However, to have a transferable method that can withstand the rigours of routine testing and consistently separate out critical variants, the platform method likely will fall short. As well as for clarification for analytical method robustness, which may still get conflated with ruggedness or reproducibility. As defined in the ICH Q2B guidelines (1), method robustness is the purposeful variation of method parameters to assess their impact. In the case of intact RP‐HPLC for proteins, the assessment of hydrophobic and hydrophilic variants is typically the method result in question. The question then becomes what method parameters should be adjusted, and what considerations need to be made.
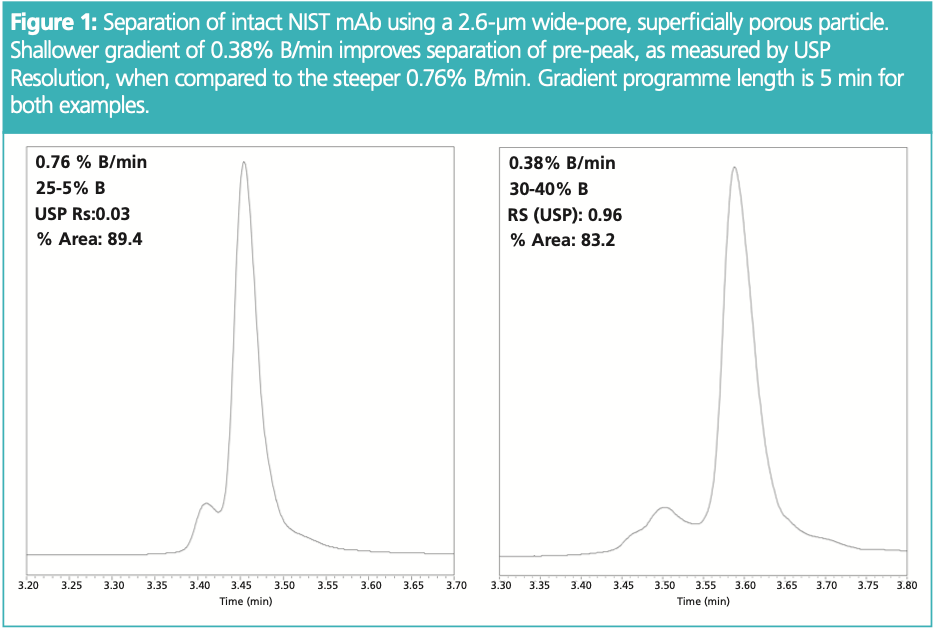
One primary method parameter to vary for method robustness assessment in any RP‐HPLC separation, intact or otherwise, is the gradient programme. That is, the time/length of the gradient programme, and how shallow the gradient slope is, as measured by a percentage of strong solvent per column volume. Gradient programme robustness assessment commonly assesses length, for example, differences in method performance between 8 min vs. 10 min gradient programmes. However, given the limitation in gradient programmes to be as short as possible to minimize on‐column degradation, the more prudent assessment is to vary starting and ending organic concentration, as this can effectively investigate gradient slope and overall method effect. Figure 1 shows the difference between 25–45% B and 30–40% B. Resolution of the pre‐peak is 0.96 USP with the shallower gradient slope, and any variability may lead to a decrease in resolution resulting in improper integration and thus misreporting of hydrophilic variants. Alternatively, flow‐rate might be modulated to demonstrate the same. It is important to note the use of superficially porous, silica based particles for this example. Wide‐pore (that is, >200 Å nominal pore diameter), superficially porous particles have been used for decades to improve the poor mass‐transfer kinetics associated to macromolecules (2). However, an undervalued aspect of wide‐pore, core–shell columns are the ability to run at faster linear velocities without an appreciable drop in chromatographic efficiency. The example in Figure 1 demonstrates separations being performed at 0.8 mL/ min using a 2.1‐mm i.d., column which represents a linear velocity of 0.4 cm/s.
For context, most intact RP‐HPLC methods are much slower; 0.1–0.2 cm/s being the most common. For clarity, the intent is not to increase throughput but to allow for shallow gradient slopes without excessively long gradient programmes.


Gradient optimization seems the most apparent for a reversed‐phase separation, however, one method development parameter that must be explored for intact RP‐HPLC is column temperature. Indeed, temperature may be the single most critical aspect of the reversed‐phase separation of mAbs and mAb fragments. Although temperature improves diffusion and mass transfer for large molecules, it has been demonstrated that high temperature is often imperative for separation of mAb and mAb fragment segments by RP‐HPLC because recovery can be impacted depending on the mAb physicochemical properties (3). Further, temperature may affect selectivity of RP‐HPLC separations of mAb fragments (4), with most methods requiring temperatures exceeding 70 °C for an optimal separation.
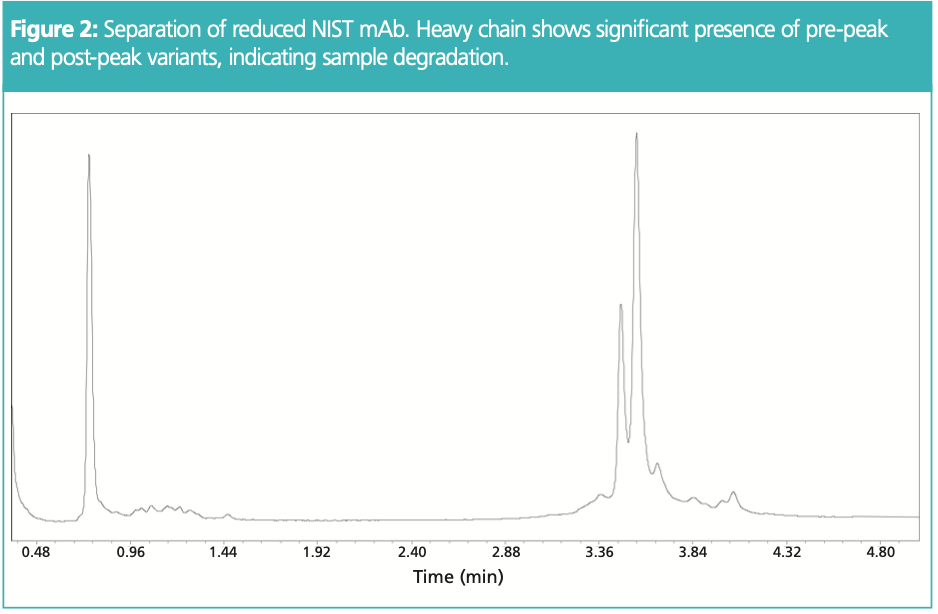
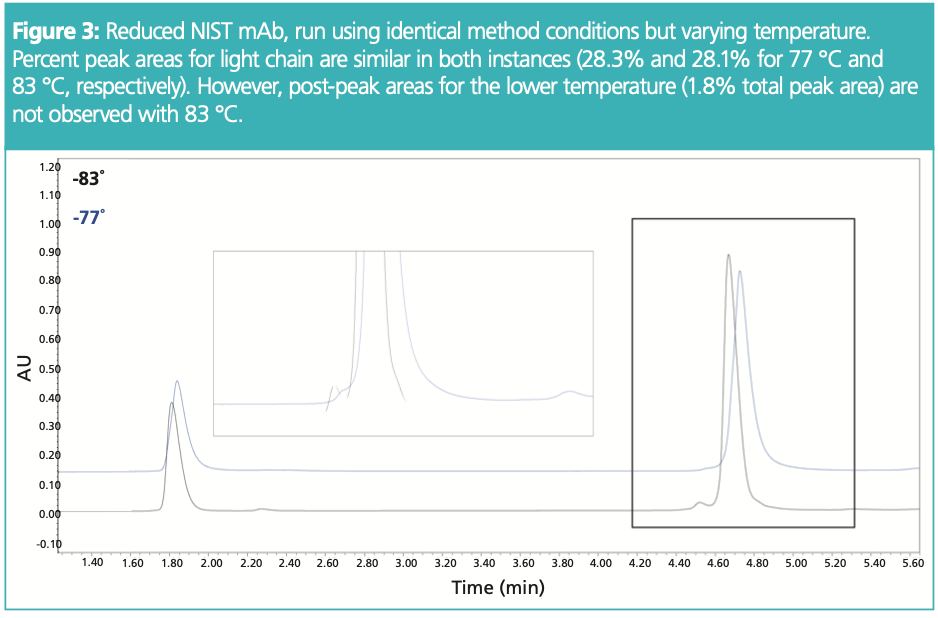
Figure 2 shows the separation of reduced NIST mAb, using a temperature of 80 °C. Being a somewhat degraded sample, it shows the level of detail that one might obtain using intact RP‐HPLC for fragments, in this instance, heavy chain and light. Figure 3 shows a different sample set, investigating the impact of purity analysis with the variation of temperature +/− 3 °C from the method starting point of 80 °C. Although the light chain shows similar % peak areas, there is some variation in pre-peak and main peak for heavy chain, with post‐peaks not being present with increased temperature. Again, since temperature can impact selectivity especially for mAb fragments, temperature should be explored not only during development for optimal selectivity but also modulated during robustness studies to assess impact to the chromatographic separation.


The next critical method parameter to evaluate for robustness would be the amount of acidic modifier added to the mobile phase. Trifluoroacetic acid (TFA) is the most common for RP‐HPLC because it is an ion‐pairing agent, acting as a counter‐ion to the positive moieties associated to proteins. This enhances the hydrophobic retention of the protein but because TFA is a strong acid, it decreases mobile phase pH thus minimizing unwanted secondary, electrostatic interactions with the inherent negative silanols present in any silica based reversed‐phase media. Further, TFA is not only critical for an optimal separation, but also for protein recovery (5).
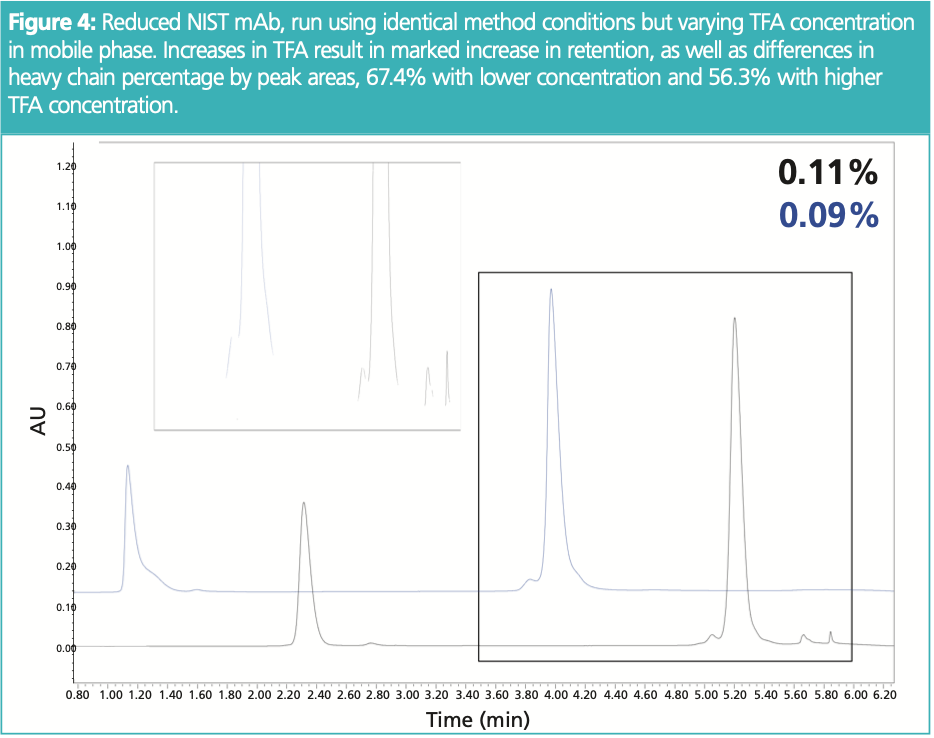
A concentration of 0.1% TFA (v/v) is commonly used for intact RP‐HPLC. This makes for facile mobile phase preparation, with the addition of 1 mL of acidic modifier to the 1 L of HPLC‐grade water and acetonitrile. However, even slight variation in the addition of TFA can have potentially drastic effects on overall retention and separation of large molecules. Although the effect of TFA concentration has mostly been studied with peptides (6), mAb fragments are subject to a similar behaviour chromatographically. That is, increases in TFA concentration can lead to overall increases in retentivity, thus changes in selectivity which must be explored during method robustness assessment. Figure 4 demonstrates the impact of TFA concentration on NIST mAb fragments, comparing method using 0.09 and 0.11% TFA in mobile phase A and B. As anticipated, there is a significant increase in retention, which can be problematic especially if the method has a retention time specification. More important though, is the variation in peak areas reported for mAb fragments. Although peak areas for light chain are similar, the heavy chain profiles vary significantly, with improved pre‐peak recovery and separation of additional, later eluting variants. The difference of 200 μL added to mobile phase seems feasible, especially given variance in pipetting technique, as well as age and quality of TFA, and so on. Further, with increases in retention and generally better separation with higher concentrations of TFA, this example demonstrates the case for implementing an expanded design of experiment wherein TFA concentration exceeds the 0.1% TFA which is commonly used. This has again been demonstrated with peptides (7) and could therefore provide utility in intact RP‐HPLC.

A final note must be made on variation of column batches. Although batch‐to‐batch performance and even intra‐batch variability should be assessed, it should be important to note what variation one might expect with intact RP‐HPLC methods. Advances in silica sol gel have certainly improved particle morphology for more consistency in particle size and pore diameter. This is especially true with the superficially porous wide‐pore particles which are becoming more common for downstream applications wherein method transferability is a priority. However, because wide‐pore particles have relatively low surface areas, sometimes as low as 25 m2/g, overall hydrophobicities between different column batches may be much more apparent. As such, one should expect a certain level of variation in overall capacity factor which may impact selectivity and resolution. This variation will then be amplified by any variance in method parameters that directly impact retentivity; namely gradient slope and TFA concentration.
Conclusion
In summary, intact RP‐HPLC is an attractive method for purity analysis. Method robustness should be assessed to ensure method transferability and sustainability, especially to support downstream applications such as quality control lot release. Specifically, the gradient programme is very important: The starting and ending concentrations of strong solvent, temperature, and acidic modifier are three critical method development parameters to explore during development but also to assess during method robustness testing.
References
1. International Conference on Harmonization (ICH) Q2B, “Validation of Analytical Procedures: Methodology”, Fed. Reg. 62(96), 27463–27467 (1997).
2. J.J. Kirkland, J. Anal. Chem. 64(11), 1239–1245 (1992).
3. S. Fekete, S. Rudaz, J.‐L. Veuthey, and D. Guillarme, J. Sep. Sci. 35(22), 3113–3123 (2012).
4. S. Fekete, S. Rudaz, J. Fekete, and D. Guillarme, J. Pharm. Biomed. Anal. 70, 158–168 (2012).
5. B. Bobály, V. Mikola, E. Sipkó, Z. Márta, and J. Fekete, J. Chrom. Sci. 53(7), 1078–1083 (2015).
6. M. Gilar, H. Xie, and A. Jaworski, J. Anal. Chem. 82(1), 265–275 (2010).
7. Y. Chen, A.R. Mehok, C.T. Mant, and R.S. Hodges, J. Chrom. A. 1043, 9–18 (2004).
Brian Rivera is the Senior Product Manager – Biologics at Phenomenex. He has worked at Phenomenex for nine years, holding other positions including technical support and sales. Prior to joining Phenomenex, Rivera worked within the biotechnology industry, including positions focused on protein purification, analytical method development, and in-process analytical support. Rivera has a bachelor’s degree from the University of California, Davis, USA.
Ivan Lebedev received his PhD in Biochemistry and Structural Biology from Stony Brook University. Since then he has been heavily involved in a variety of protein/enzyme-, oligo-, and bioconjugation-related work at Phenomenex and previously ReadCoor (now part of 10x Genomics). He is currently an Application Scientist at Phenomenex focusing on expanding the portfolio of biological applications and addressing unique customer needs.
Chad Eichman is the Global Business Unit Manager – Biopharmaceuticals at Phenomenex, where he develops and leads the strategic plans for Phenomenex’s biopharmaceutical business. He received his B.S. in chemistry from the University of Wisconsin–Madison (USA) and his Ph.D. in organic chemistry from The Ohio State University (USA). After a postdoctoral appointment at Northwestern University (USA) and an assistant professorship at Loyola University Chicago (USA), Eichman joined Phenomenex in 2017.
E-mail: BrianR@Phenomenex.com
Website: www.phenomenex.com/biozen





















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.



Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

