A Mass Spectrometry Primer: Part I
LCGC North America
Michael Balogh begins a three-part overview series, answering questions about mass spectrometers, their use and capabilities, and including links to more detailed further reading in readily available articles written for the nonspecialist.
Technological changes affecting how we access information — both in the depth of knowledge we can access and the speed — leads to some observations about electronic versus print media. First, anything committed to public view must be of high scholarly order, often serving as a prime resource for equations and things we can't or won't remember. Second, once words have been printed, the value of the meaning they impart decreases as new understanding takes shape.



The electronic communication marvels still fall short of resolving all of the static shortcomings of print: increasingly ubiquitous blogs typically are narrowly focused, being written by a single individual, and the benefit of an interactive conversation developing deeper understanding is still on the horizon.
Therein lays the premise for this project. Primers in various forms can be found from a variety of authors and many of them are referenced for further reading in this one. What makes this one different is its continually self-validating existence residing on the web. Thousands of elite programmers at Google have developed uncanny heuristic algorithms to categorize and rate sites for quality. That is, searching for a mass spectrometry (MS)-related term will not turn up a site deemed "commercial" as a very high "hit."
Readers of this column are familiar with the extent to which the editors go to ensure the technology is examined in an open "vendor neutral" fashion. The electronic primer, unfolding in print over the remainder of 2008, will reside on the Waters website and will be updated as readers' comments and suggestions come in either from readers in the U.S. and Europe or after visiting the Waters website (see "Resource Library," "Primers" at www.waters.com).


Who Uses MS?
This primer covers a wide range of topics related to modern MS practices and answers some frequently asked questions about the use and capabilities of mass spectrometers. Links also are provided to articles written for nonspecialists for more in-depth reading. The first section examines who uses mass spectrometers followed by how compounds are ionized in the source to be analyzed by mass spectrometers. A description of the various types of mass spectrometers is next and a discussion of the important topics of mass accuracy and resolution — or how well we can tell differences between closely related compounds. Chemistry, sample preparation, and data handling are considered, as well as the definition of some terms commonly in use in the most prevalent forms of MS practice today.
Before considering MS, you should consider the types of analyses you perform and the kind of results you expect from them:
- Do you want to analyze large molecules, like proteins and peptides, or acquire small, aqueous-molecule data?
- Do you look for target compounds at a determined level of detail, or do you want to characterize unknown samples?
- Are your current separations robust, or must you develop methods from complex matrixes?
- Do you require unit mass accuracy — say, 400 MW — or accuracy to 5?ppm, as in 400.0125 MW (or 2 mDa at mass 400)?
- Do you process hundreds of samples a day? Thousands? Tens of thousands?
Who Uses MS?
Researchers and practitioners from various disciplines and subdisciplines within chemistry, biochemistry, and physics regularly depend upon MS analysis. Pharmaceutical industry workers involved in drug discovery and development rely on the specificity, dynamic range, and sensitivity of MS to differentiate closely related metabolites in a complex matrix and, thus, identify and quantify metabolites. Particularly in drug discovery, where compound identification and purity from synthesis and early pharmacokinetics are determined, MS has proved indispensable.
Biochemists expand the use of MS to protein, peptide, and oligonucleotide analysis. Using mass spectrometers, they monitor enzyme reactions, confirm amino acid sequences, and identify large proteins from databases that include samples derived from proteolytic fragments. They also monitor protein folding, carried out by means of hydrogen–deuterium exchange studies, and important protein–ligand complex formation under physiological conditions.


Figure 1: Singly and doubly charged ions of Substance P. The inset shows a closer view of the appearance in the spectral display for singly and doubly charged species.
Clinical chemists, too, are adopting MS, replacing the less-certain results of immunoassays for drug testing and neonatal screening. So are food safety and environmental researchers. They and their allied industrial counterparts have turned to MS for some of the same reasons: PAH and PCB analysis, water quality studies, and to measure pesticide residues in foods. Determining oil composition, a complex and costly prospect, fueled the development of some of the earliest mass spectrometers and continues to drive significant advances in the technology.
Today, the MS practitioner can choose among a range of ionization techniques that have become robust and trustworthy on a variety of instruments with demonstrated capabilities.
What are mass spectrometers, and how do they work?
Mass spectrometers can be smaller than a coin, or they can fill very large rooms. Although the various instrument types serve in vastly different applications, they nevertheless share certain operating fundamentals. The unit of measure has become the dalton (Da), displacing other terms such as amu. 1 Da = 1/12 of the mass of a single atom of the isotope of carbon-12 (12 C).
Once employed strictly as qualitative devices — adjuncts in determining compound identity — mass spectrometers were once considered incapable of rigorous quantitation. But in more recent times, they have proved themselves as both qualitative and quantitative instruments. A mass spectrometer can measure the mass of a molecule only after it converts the molecule to a gas-phase ion. To do so, it imparts an electrical charge to molecules and converts the resultant flux of electrically charged ions into a proportional electrical current that a data system then reads. The data system converts the current to digital information, displaying it as a mass spectrum.


Figure 2: Multiply charged, large molecules (27 to 46 charges shown for human transferrin) with measured mass (79,550 Da) indicated by the maximum entropy algorithm allows measurement of masses well in excess of the design range of the instrument with great accuracy.
Ions can be created in a number of ways suited to the target analyte in question: some examples — by laser ablation of a compound dissolved in a matrix on a planar surface such as by MALDI, by interaction with an energized particle or electron such as in EI, or as part of the transport process itself as we have come to know ESI, in which the eluent from a liquid chromatograph receives a high voltage, resulting in ions from an aerosol.
The ions are separated, detected, and measured according to their mass-to-charge ratios (m/z). Relative ion current (signal) is plotted versus m/z, producing a mass spectrum. Small molecules typically exhibit only a single charge: the m/z is therefore some mass (m) over 1. The "1" being a proton added in the ionization process (represented M+H+ or M- H+ if formed by the loss of a proton) or if the ion is formed by loss of an electron, it is represented as the radical cation (M+ ). The accuracy of a mass spectrometer or how well it can measure the actual true mass can vary, as will be seen in later sections of this primer.
Larger molecules capture charges in more than one location within their structure. Small peptides typically can have two charges (M+2H+ ), while very large molecules have numerous sites, allowing simple algorithms to deduce the mass of the ion represented in the spectrum.
Figure 1 shows singly and doubly charged ions of Substance P. The inset shows a closer view of the appearance in the spectral display for singly and doubly charged species.
Multiply charged, large molecules (27 to 46 charges shown for human transferrin) with measured mass (79,550 Da) indicated by the maximum entropy algorithm allows measurement of masses well in excess of the design range of the instrument with great accuracy (Figure 2).
How large a molecule can I analyze? Desorption methods (described in this primer) have extended the ability to analyze large, nonvolatile, fragile molecules. Routine detection of 40,000 Da within 0.01% accuracy (or within 4 Da) allows the determination of minor changes such as posttranslational modifications of proteins. Multiple charging extends the range of the mass spectrometer well beyond its designed upper limit to include masses of 1,000,000 Da or more.
Isotope and elemental MS: Natural isotope abundance is well characterized. Though often thought to be stable, it nevertheless can display significant and characteristic variances. Isotope ratio measurements are used in metabolic studies (isotope-enriched elements serve as tracers) and also in climatic studies that measure temperature-dependent oxygen and carbon changes. In practice, complex molecules are reduced to simple molecular components before being measured using high-accuracy capabilities such as those found on magnetic sector instruments (see the following section).


See MS â The Practical Art, LCGC (chromatographyonline.com)
Elemental analysis typically is performed on inorganic materials — to determine elemental makeup, not structure — in some cases using solid metal samples. Inductively coupled plasma (ICP) sources are common where a discharge (or lower power-glow discharge) device ionizes the sample. Detection using dedicated instruments, at the parts-per-trillion level, is not uncommon.
The Analyzer: the heart of a mass spectrometer: The analyzer is an instrument's means of separating or differentiating introduced ions. Both positive and negative ions (as well as uncharged, neutral species) form in the ion source. However, only one polarity is recorded at a given moment. Modern instruments can switch polarities in milliseconds, yielding high-fidelity records even of fast, transient events like those typical of ultrahigh-pressure liquid chromatography (UHPLC) or GC separations in which peaks are only about 1-s wide.
Electron Ionization (EI)
Many are familiar with EI. (Sometimes the earlier phrase "electron impact" is used — although, technically, it is incorrect). EI, often performed by exposing a sample to 70 eV electrons, is referred to as a "hard" technique. The energy of the electrons interacting with the molecule of interest generally is much greater than that contained in its bonds, so ionization occurs. The excess energy breaks bonds in a well-characterized way. The result is predictable, identifiable fragments from which we can deduce the molecule's identity. Abstraction of only an electron from the outer shell yields a radical cation in the positive mode (M+ ) while the additional energy internalized creates a rich spectrum of fragments. Unlike "softer" atmospheric ionization techniques that produce a spectral response sometimes characteristic of the manufacturer's particular source design, the EI technique is fairly independent of the source design. A spectrum produced by one EI instrument looks much like a spectrum of the same compound from another EI instrument, a fact that lends itself to creating spectral libraries to match unknowns to reference spectra.

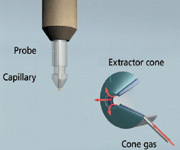
Figure 3: Simplified schematic showing an ESI probe in a typical position in front and orthogonal to the MS ion inlet (red arrows show flow of nitrogen counter-current gas, which improves desolvation and reduces clusters appearing as fragments in spectra).
Chemical Ionization (CI)
Molecules that fragment excessively call for "soft" techniques. Chemical ionization (CI) produces ions by a gentler proton transfer process that preserves and promotes the appearance of the molecular ion itself. The sample is exposed to an excess of reagent gas such as the gas that evolves when methane forms the protonated molecular ion (M+H). The reverse process can produce negative ions. Transferring the proton to the gas molecule can, in some cases, produce the negative ion (M-H).
CI is sometimes used for compounds with chemistry similar to those analyzed by EI to enhance the abundance or appearance of the molecular ion in favor of significant fragmentation. Similar to EI, samples must be thermally stable because heating in the source causes vaporization. The ionization mechanism of CI relies on EI for the initial ionization step but within the source is a chemical reagent gas, such as methane, butane or ammonia, at high pressure. The reagent gas, which is present at a much higher concentration than the analyte (R), is ionized by electron ionization to give primary R1{ reagent ions. The collision of the R1{ ions with neutral R molecules lead to the formation of stable secondary ions which are the reactant species which then ionize analyte molecules (A) by ion–molecule reactions.
For example, the ion–molecule reaction between a methane ion and a methane molecule gives rise to the fairly stable CH5+ species.
The reactant ion CH5+ can ionize neutral analyte molecules (A) by proton transfer, hydride abstraction, or charge exchange.
The most common ionization reactions are protonation, which is favored for molecules with proton affinities higher than the reagent. Hydride abstraction is common for lower proton affinity molecules and charge exchange occurs with reagents of high ionization energy.
The substance to be analyzed is at a much lower pressure than the reagent gas. If we consider methane as the reagent gas, the electron impact causes mainly ionization of the methane. This fragments in part to CH3+ . These species then undergo ion molecule reactions under the high source pressures employed.
CH5+ can act as a Bronsted acid and C2H5+ as a Lewis acid to produce ions from the analyte.
Careful choice of the CI reagent gas can improve charge transfer to an analyte molecule as the gas phase acidity of the chemical ionization gas influences the efficiency of the charge transfer. In CI, the analyte is more likely to result in a molecular ion with the reduced fragmentation conserving the energy normally internalized in EI to break bonds.
Negative Chemical Ionization (NCI)
A variation, negative chemical ionization (NCI), can be performed with an analyte that contains electron-capturing moieties (for example, fluorine atoms or nitrobenzyl groups). Sensitivity can be increased to as much as 100 to 1000 times greater than that of EI. NCI is applicable to a wide variety of small molecules that are (or can be) chemically modified to promote electron capture.
In negative ion, there are two primary mechanisms whereby negative ions are produced: electron capture and reactant ion chemical ionization. Under CI conditions, electronegative molecules can capture thermal electrons to generate negative ions. True negative ion chemical ionization occurs by reaction of an analyte compound (AH) with negatively charged reactant ions (R- or R- ). Several types of ion–molecule reactions can occur, the most common being proton abstraction.


See MS â The Practical Art, LCGC (chromatographyonline.com)
As the proton affinity (basicity) of the reactant ion increases, the more likely proton abstraction is to occur.
Fragmentation
Collisionally induced dissociation (CID), also referred to as collisionally activated dissociation (CAD), is a mechanism by which molecular ions are fragmented in the gas phase, usually by acceleration by electrical potential to a high kinetic energy in the vacuum region followed by collision with neutral gas molecules such as helium, nitrogen, or argon.
A portion of the kinetic energy is converted or internalized by the collision, which results in chemical bonds breaking, and the molecular ion is reduced to smaller fragments. Some similar "special purpose" fragmentation methods include electron transfer dissociation (ETD) and electron-capture dissociation (ECD). See the section on "Biomolecular ionization methods."
Gas Chromatography (GC)
Perhaps the first encounter with a mass spectrometer for many is as the detector for a gas chromatograph. The range of GC–MS instrument types has expanded to transcend the limitations of earlier instrument designs or to meet increasingly stringent legislation in applications like environmental analysis, food safety screening, metabolomics, and clinical applications like forensics, toxicology, and drug screening.
In the past, two types of mass spectrometers dominated GC–MS analysis: magnetic sector and the single-quadrupole instruments. The former, which offered high resolution and accurate-mass analyses, was used in applications that required extreme sensitivity. The latter performed routine analysis of target compounds.
The most challenging GC–MS analyses were reserved for magnetic sector instruments: dioxins in environmental–industrial samples or screening for the illegal use of performance-enhancing drugs in competitive sports. Femtogram detection levels, at high resolution and selectivity are achieved easily on magnetic sector instruments.
Shortly after their introduction, quadrupole GC–MS systems gained acceptance in target analysis applications. U.S. EPA methods dictated the use of quadrupole GC–MS instruments to analyze samples for numerous environmental contaminants. Because those applications require only picogram-to-nanogram levels of detection, the poorer sensitivity of the quad relative to the sector was not a limitation. On the contrary, the greatly reduced cost, ease-of-use, and portability proved a godsend.
Liquid Chromatography (LC)
The revolutionary technology that gave us analytical access to about 80% of the chemical universe unreachable by GC also is responsible for the phenomenal growth and interest in MS in recent decades. A few individuals are singled out (see the section on "A Brief History") for coupling LC with MS. Beginning arguably in the 1970s, LC–MS as we know it today reached maturation in the early 1990s. Many of the devices and techniques we use today in practice are drawn directly from that time.

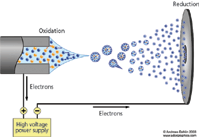
Figure 4: After formation the ions are "dragged" through a potential gradient (an electric field) to the counter plate. (Figure used by permission from Andreas Dahlin www.adorgraphics.com.)
LC was defined in the early 1900s by the work of the Russian botanist, Mikhail S. Tswett. His studies focused on separating leaf pigments extracted from plants using a solvent in a column packed with particles. In its simplest form, LC relies on the ability to predict and reproduce with great precision competing interactions between analytes in solution (the mobile or condensed phase) being passed over a bed of packed particles (the stationary phase). Development of columns packed with a variety of functional moieties in recent years and the solvent delivery systems able to precisely deliver the mobile phase has enabled LC to become the analytical backbone for many industries.
The abbreviation HPLC was coined by Csaba Horváth in 1970 to indicate that high pressure was used to generate the flow required for LC in packed columns. Continued advances in performance since then, including development of smaller particles and greater selectivity also saw the definition of the abbreviation change to high performance liquid chromatography.
In 2004, further advances in instrumentation and column technology achieved significant increases in resolution, speed, and sensitivity in LC. Columns with smaller particles (1.7 μm) and instrumentation with specialized capabilities designed to deliver mobile phase at 15,000 psi (1000 bar) came to be known as UHPLC technology, representing the differentiated term ultrahigh-pressure LC. Much of what is embodied in this current technology was predicted by investigators such as Sir John Knox in the 1970s. Knox predicted the optimum particle diameters would be 1–2 μm and chromatography would be thermally sensitive to frictional heat. Technology capable of developing robust, uniform small particles was necessarily encountered and resolved on the path to developing UHPLC for widespread use. A good basic primer on HPLC and UHPLC can be seen at www.waters.com <Resource Library>.
Electrospray (ESI)
The general term "atmospheric pressure ionization" (API) includes the most notable technique, ESI, which itself provides the basis for various, related techniques capable of creating ions at atmospheric pressure rather than in a vacuum. The sample is dissolved in a polar solvent (typically less volatile than that used with GC) and pumped through a stainless steel capillary that carries between 2000 and 4000 V. The liquid aerosolizes as it exits the capillary at atmospheric pressure, and the desolvating droplets shed ions that flow into the mass spectrometer induced by the combined effects of electrostatic attraction and vacuum.
Figure 3 is a simplified schematic showing an ESI probe in a typical position in front and orthogonal to the MS ion inlet.
A cone or countercurrent gas often is applied to aid desolvation of liquid droplets as they enter the rarified gas vacuum region of the analyzer.
The mechanism by which potential transfers from the liquid to the analyte, creating ions, remains a topic of controversy. In 1968, Malcolm Dole first proposed the charge residue mechanism in which he hypothesized that as a droplet evaporates, its charge remains unchanged. The droplet's surface tension, ultimately unable to oppose the repulsive forces from the imposed charge, explodes into many smaller droplets. These Coulombic fissions occur until droplets containing a single analyte ion remain. When the solvent evaporates from the last droplet, a gas-phase ion forms.
In 1976, Iribarne and Thomson proposed a different model, the ion evaporation mechanism, in which small droplets form by Coulombic fission, similar to the way they form in Dole's model. However, according to ion evaporation theory, the electric field strength at the surface of the droplet is high enough to make leaving the droplet surface and transferring directly into the gas phase energetically favorable for solvated ions.
It is possible that the two mechanisms might actually work in concert: the charge residue mechanism dominant for masses higher than 3000 Da while ion evaporation dominant for lower masses (see R. Cole, J. Mass Spec. 35, 763–772 [2000]).
The liquid from the liquid chromatograph enters the ESI probe in a state of charge balance. So when the solvent leaves the ESI probe it carries a net ionic charge. To ensure that ESI is a continuous technique, the solution must be charged by electrochemical reactions, whereby electrons transfer to a conductive surface acting as an electrode. Among other effects, this process can lead to pH changes. It is assumed that, in positive mode, positive-charged droplets leave the spray, and electrons are accepted by the electrode (oxidation). (The reverse would be true in negative mode.) The surface area of the electroactive electrode, the magnitude of the current, and the nature of the chemical species and their electrode potentials all exert an effect.
Over all, ESI is an efficient process. However, the activation energy and energy difference for the reaction, in total, for individual species varies. The flow rate of the solution and the applied current define limits for each droplet. Competition between molecules occurs, and suppression of analytes of interest is not uncommon.
After formation, the ions are "dragged" through a potential gradient (an electric field) to the counter plate (Figure 4).
Extensions of basic ESI theory, such as reducing the liquid to extremely low volumes — for example to 30 nL/min in the case of nanospray — have proved effective, especially in sample-limited studies of proteins and amino acids.
Atmospheric Pressure Chemical Ionization (APCI)
Although work demonstrating APCI was published in parallel with that demonstrating ESI, APCI was not widely adopted until ESI was commercialized, which occurred in the wake of Fenn's work in 1985.
Horning first introduced APCI in 1973 to analyze volatile compounds using various introduction techniques, one of which was HPLC. The adjunctive capability of APCI permits analytes that resist conversion to gas-phase ions by ESI, the less polar and more volatile ones introduced into a mass spectrometer from a condensed phase, or liquid, stream. Unlike ESI, APCI transfers neutral analytes into the gas phase by vaporizing the introduced liquid in a heated gas stream. Chemical ionization relies on the transfer of charged species between a reagent ion and a target molecule to produce a target ion that can be mass analyzed. Most commonly, in positive ion mode, an adduct forms between the target molecule and the small H+ ion, although adducts with salts are common as well. For example, the ammonium adduct can form (M+NH4)+ when the weak-acid–weak-base salt, ammonium acetate, is present in the mobile phase, a modifier often used in place of the less volatile and highly ionic phosphate buffer. At higher salt concentrations, competition between the protonated and ammoniated forms can produce a decreased response for both. The maximum number of ions capable of forming by APCI is much greater than it is in ESI because reagent ions form redundantly. The liquid is pushed through a nonconductive tube, usually of fused-silica glass, around which a nebulizing gas flows. The resultant fine droplets collide with the inner, heated wall of a tube or probe that extends beyond the end of the nonconductive tube and are, thus, converted to the gas phase. This ionization type often is performed at much greater linear velocities than those of the HPLC or UHPLC flow rates normally associated with electrospray. Contemporary instruments, however, provide much greater desolvation capacities, enhancing performance for all aerosol-dependent techniques.
The desolvated analyte molecules are then ionized via CI. The ionizing potential is applied, not through the liquid as in ESI, but at the tip of a needle as a plasma, or corona, through which the droplets pass. In effect, the mobile phase acts as an intermediary transferring the charge to the analyte. Hence, the early name given APCI: "solvent-mediated electrospray."
Biomolecular Ionization Methods
Ionization techniques have been developed to aid identification of biomolecules rather than aggressively reduce the molecule to components. Two "energy deposition" processes, electron capture dissociation (ECD) (R.A. Zubarev, Curr. Opin. Biotechnol. 15, 12–16 [2004]) and electron-transfer dissociation (ETD) (J.J. Coon, J. Shabanowitz, D.F. Hunt, and J.E. Syka, J. Am. Soc. Mass Spectrom. 16, 880–882 [2005]) are commonly recognized in biomolecular analysis and proteomics. Both cleave bonds adjacent to sites of electron capture and unlike other fragmentation processes, such as CID, the cleaved bonds are not the most labile within the molecule. The cleavages observed are less dependent upon the peptide sequence, so cleavages between most amino acids in the peptide backbone tend to be independent of the molecule's size. The dominant fragmentation in ECD and ETD of peptides is the formation of c and z· ions. ECD has been demonstrated to be useful for the analysis of labile post-translational modifications such as phosphorylation and O-glycosylation and for fragmentation analysis of intact proteins.
It has been shown that for elucidating structural details of proteins in solution, ESI-MS is aided further awhen coupled with amide hydrogen/deuterium (H/D) exchange analysis. Charge-state distributions, and the envelopes of charges ESI forms on proteins, can provide information on solution conformations of larger proteins with smaller amounts of sample not easily performed using other techniques such as near ultraviolet circular dichroism (CD) and tryptophan fluorescence (however, typically it is used in conjunction with these techniques and others such as nuclear magnetic resonance). The other techniques measure the average properties of large populations of proteins in solution so an additional advantage seen with MS is its ability to provide structural details on transient or folding intermediates.
Alternative ionization means: Pure or neat compounds can be introduced into the ion source, having been deposited in the tip of a rod or solids probe. With heat, the sample sublimates or evaporates into the gas phase. In most cases, ionization follows by the means described here. But in some cases, ionization occurs simultaneous with the sublimation or evaporation.
- APPI
–Direct or dopant-assisted photon ionization of analytes with ionization potential below 10 eV (the primary photon energy output by a krypton gas lamp). The ionization potential for solvents commonly used in LC are above 10 eV. APPI is one of the primary API alternatives because it extends the ionization range to more nonpolar analytes than either ESI or APCI can ionize.
- MALDI
–Soft ionization for intact proteins, peptides, and most other biomolecules (oligonucleotides, carbohydrates, natural products, and lipids) and analysis of heterogeneous samples (analysis of complex biological samples such as proteolytic digests)
–High-energy photons interact with a sample embedded in an organic matrix typically with subpicomole sensitivity.
–First introduced in 1988 by Tanaka, Karas, and Hillenkamp.
- FAB
–An early form of soft ionization using a stream of cesium ions to "sputter" ions from a sample dissolved in a glycerol, or similar, matrix
- Desorption
–Plasma desorption nuclear fission fragments interact with a solid sample deposited on a metal foil
–Secondary ion MS (SIMS) high- velocity ions impact a thin film of sample deposited on a metal plate or contained in a liquid matrix (liquid SIMS).
–Field desorption: a high field gradient is imposed on a sample deposited on a support
–Desorption ESI (DESI): along with closely related techniques like DART (direct analysis real time), ASAP (atmospheric solids analysis probe) and others recently introduced to the market these tend to create ions secondary to some interaction on a surface. In DESI, an energized liquid stream is aimed at a sample deposited in a flat surface, causing secondary ionization to occur at atmospheric pressures.
Upcoming installments of "MS – The Practical Art" will continue the discussion of MS basics. Part II of "A Mass Spectrometry Primer" will provide a brief history of MS and will describe the types of instruments currently in use. Part III will discuss mass accuracy and resolution, how to interpret MS output, quantitation and calibration, and solvents and caveats for LC–MS.
Acknowledgment
In view of the wide range of material drawn upon and the comprehensive nature of a project such as this, the advice and assistance of a number of people with equally diverse and accomplished backgrounds is indispensable. One in particular deserves notice and my thanks: Hilary Major in Manchester, UK for her contributions.

Michael P. Balogh
Michael P. Balogh
"MS — The Practical Art" Editor Michael P. Balogh is principal scientist, MS technology development, at Waters Corp. (Milford, Massachusetts); a former adjunct professor and visiting scientist at Roger Williams University (Bristol, Rhode Island); cofounder and current president of the Society for Small Molecule Science (CoSMoS) and a member of LCGC's editorial advisory board













Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.





