Linear Thinking in a Nonlinear World
LCGC North America
A very useful construct in engineering and analytical chemistry is the concept of a time-invariant linear system. The properties of the system (as for a mathematical function) are defined by a relationship between the response and the excitation, and ...
A very useful construct in engineering and analytical chemistry is the concept of a time-invariant linear system. A system can be defined as an equation, a circuit, a transducer, an instrument, an analytical method, or a mammal, among others. A system can be probed with a signal (an input, a voltage, a concentration, a frequency, a mass, a dose, and so forth). In engineering jargon, we refer to an excitation, which might be an impulse, a pulse of finite width or a continuous stress. The properties of the system (as for a mathematical function) are defined by a relationship between the response and the excitation. The two properties that define a linear system are that the response is directly proportional to the excitation amplitude (Figure 1) and that superposition holds (Figure 2) meaning that the response to a combination of excitations is equal to the sum of their individual response functions when applied alone. While a system can behave linearly at one time and not at another, we all prefer, or at least find comfort in, time-invariant systems that are linear (Figure 3).
Given that this is a chromatography publication, let us interpret what we mean in chromatographic terms. A linear chromatograph is one where the scale of the response peak (height and area) is directly proportional to the amount of material placed on the column. Superposition then demands that the response to any given component is completely independent of all other components injected in the sample. In other words, compounds in a mixture behave exactly as they would individually. We want to be very clear that we are not specifically referring here to analytes, but rather to all components loaded on the column whether to be determined or not. We have made an impulse excitation of the column when the volume injected is small compared to the peak volume for the narrowest peak that is eluted. This is the assumption for nearly all analytical chromatography applications but is not the case for many preparative separations.



We can define the system in a variety of ways. For example, the column is a system on which we impose concentration profiles in the mobile phase as a function of time (or volume) and we measure concentration profiles as a function of time (or volume) at the distal end. We can just as well view the combination of the autosampler, column, detectors, and data processing as the system. As John Dolan has told us for decades, to understand the complete instrument, it is helpful to understand its individual components. This is especially helpful when troubleshooting, although the system can be more than the sum of its parts.
Ultimately, we are asked to define a method and, thus, we add a sample preparation scheme and go through various procedures to establish that the method is valid for the intended purpose. To sensibly do that we must understand the purpose, which (for many) will be to "get a number," but that is provincial thinking. The real purpose is inevitably to make a decision in response to a question: "Can this production lot be released for shipping?" "Did the athlete use a steroid?" "Is the food fresh?" "Does Mr. Jones have a disease?" "Is the generic drug bioequivalent to the innovator's formulation?"

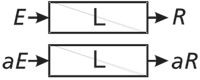
Figure 1: For a linear system L, the amplitude of the response R is directly proportional to the amplitude of the excitation E or stimulus.
Our personal experience is nearly exclusively in "blood and guts" bioanalytical chemistry. Over the years it became very clear that it is only quite rare that an analytical method behaves linearly and if it does so, the circumstances are often quite limited. A result of this problem was the development of a method validation formality that evolved in the mid-1990s in a consortium fashion across commercial, academic and government laboratories. With the rapid rise of outsourcing analytical chemistry, government agencies and sponsors of research wanted to be sure the numbers they obtained were good numbers, meaning numbers that could be trusted to make a decision. Given our experience, we limit this column installment to preclinical and clinical samples derived from mammals. Figure 4 provides a systems view of a mammal. It is a fair statement that practically nothing about a mammal behaves linearly.
Only by combining the properties of the mammal as a system with those of the method as a system, can we be sure the numbers are good for their purpose. The goal of this article is to not review the methods validation literature for bioanalytical chemistry. There are a number of reviews which do that, especially well for single analyte assays. It is very important to distinquish method validation for products and raw materials from that for bioanalytical assays. There are two essential citations for bioanalysis (1,2).
We are rather dismayed, however, by aspects of obtaining and handling the samples that can foil the intent of the finest methods development effort. While many of these will seem obvious, only rarely are they considered by bioanalytical chemists. Thus, our purpose is to stimulate thought. We believe linear systems thinking is a useful way to be reminded of our nonlinear world.

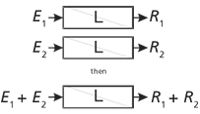
Figure 2: Superposition principle: For a linear system L, the responses to a combination of stimuli are the sum of the responses to the stimuli applied individually.
Determining One Or Few Analytes In Mammalian Systems
It is often what you can not see that hurts you, especially with selective methods. The more selective a method becomes, the more likely we are to be fooled when the superposition principle fails us. Time and again we have seen "undetectable" components in large relative amounts that damage the response to an analyte signal from a mass spectrometer, or earlier electrochemical, ultraviolet absorbance, or fluorescence liquid chromatography (LC) detectors. We talk about the biological matrix (also known as "everything except the analytes") when we should be keeping in mind the variable biological matrix. The factors of variability are species to species, individual to individual (no two of a given species are alike), diet to diet, time to time and dose to dose. Age, sex, and time of day can either make huge differences or rather small ones and it is not clear a priori which will be the case. The very act of collecting a sample can change both the analytes of interest and the matrix. Decades ago we would develop methods in "pooled samples from healthy subjects" and would joke about swimming in the urine pool. We would "spike" the pool with controlled amounts of analyte and then describe linearity, absolute and relative recovery for extractions, lower and upper limits of quantitation, accuracy, and precision. We would explore probable interferences and salute the "clean baseline" when the analytes of interest were not present. After all of this, it was not uncommon to get a less than satisfactory number. Why? It was often because what you can not see can hurt you. Today, we speak of incurred samples to distinguish samples that have passed though a mammal from those contrived in the laboratory to develop a method. Surprising things happen in mammals and the FDA has made this topic a priority, but only very recently.

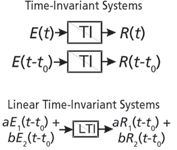
Figure 3: A time-invariant linear system (or method) is what we desire, but this is rarely the case and usually over very limited spans of time and stimulus amplitude.
Science is about curiosity. Years back we learned of analytical chemists who processed the samples that came in and sent the results out and had no idea where the samples came from or how the data would be interpreted. This shocked us. We concluded that they were chemists, but not scientists. In many cases, this could not be helped because the sponsor of the work thought that analytical chemistry was a black box (a linear system?) where a sample went in one side and a number came out the other. This can be described as the industrialization of analytical chemistry. It can work, but there are risks.
Here is a list of a few confounding biological issues for mammalian bioanalytical chemistry:
- Diet
- Postprandial dosing and sampling time (food matters — what, when, and how much?)
- Incurred samples vs. laboratory spiked biomatrix
- Conjugation before and after metabolism
- Diurnal variations
- Influence of physical and emotional stress (for example, picking up a rat, waking a baby, or meeting a person in a white coat)
- Multiple drugs interacting where the presence of one inhibited an enzyme or transporter acting on the other
- Disease alters the matrix — blood from a late-stage cancer patient on 12 medications is not the blood you used to develop and validate your method.
For humans, these topics can largely be controlled if we are alert and carefully document the protocol. The unique aspect of human mammals is that we can give them instructions and ask them for data such as "How do you feel?" or "What did you have for breakfast?" We are all familiar with the instruction to drink nothing but clear liquids after 8 p.m., eat no breakfast and take no drugs before your appointment for an 8 a.m. blood draw. For animals, the challenge is greater, especially the influence of stress associated with human handling which can dramatically influence both chemical and physical markers. The physical markers, of course, include blood pressure, heart rate, ECG, EEG, and so forth. Actually it can be even more subtle in that a human walking into an animal facility can create certain anxieties for a rat, not unlike those experienced by Indiana Jones in the presence of a snake. More than 15 years ago this problem was made evident by microdialysis sampling of central neurochemical events in awake freely moving rodents. About seven years ago the problem also became quite evident for endogenous markers like circulating corticosterone, epinephrine, and norepinephrine, and for pharmacokinetic studies where the very act of obtaining a blood sample changed the data that was sought. Stress alters chemistry and the chemistry then alters the routing of blood flow, the heart rate, and the availability of glucose to muscles. To avoid the stress response associated with handling an animal, or even being in the room with them, a new approach was developed that enabled programmed dosing and subsequent automated collection of blood samples, microdialysates and physiological data. This was accomplished with a movement responsive caging system that enables multiple connections between a rodent and the outside world of sampling and measuring (3). Figure 5 illustrates one of several configurations for rodents. The same approach has been used on a larger scale for pigs, a far better model for human physiology and chemistry. Pigs also enable the use of solid dosage forms at the potency designed for humans; an impossibility with rodents. When these automated tools are used, the data are far tighter on both the y axis and the time axis. Collecting serial information in the same animal over a number of days enables an animal to be its own control with respect to changes in dose. Not long ago, concentration data at one time point meant one mouse was sacrificed per data point. This can now be avoided. Multiple pharmacokinetic curves can be obtained over time in a single rat or mouse, painlessly.

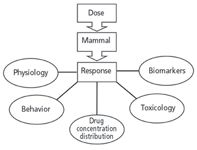
Figure 4: Considering a mammal as a system and a drug dose as a stimulus, the responses of interest cover a range of scientific disciplines. Reality for biology is not linear.
Determining Many Analytes In Mammalian Systems ("-omics")
Once again we are fascinated by the prospects of metabolic profiling, a very hot topic in the early 1970s that quickly faded when practically nothing was learned from it. As with many things, we have given it new names, among them proteomics, peptidomics, glycomics, and metabolomics. We have dramatically improved tools. Long gone is the notion of profiling 12–24 h human urine samples using ion-exchange chromatography, which itself took 24 h per sample. The hypothesis was clear: something new and useful would certainly be found that correlated with disease. As a rule, all that was found were peaks, lots and lots of peaks.
There was somewhat greater success with capillary gas chromatography, at least in terms of understanding inborn errors of metabolism in genetic diseases, often involving lipids. Today, virtually every state screens all neonates to detect these metabolic disorders with mass spectrometry tools and in some cases this has allowed for dietary changes with great therapeutic benefit to the children.
The -omics idea is the same one we had 40 years ago. The hope is that new higher resolution tools will help us find new surrogate markers of disease, including xenobiotic induced toxicity. Will it work this time around? There is no doubt that we can now look in the weeds with modern multidimensional nuclear magnetic resonance (NMR) and far higher resolution chromatographies coupled to mass spectrometry. We have orders of magnitude more speed and far lower limits of quantitation augmented by amazing automation tools in the form of robotics and informatics. We also think of systems (that word again!) biology and believe that networks of metabolites will reveal more than any individual signal. We like the concept, but see dangers in science that seems to follow the same theory that enough monkeys typing will eventually write Shakespeare's plays. We believe a hypothesis-driven focus on metabolites of known structure linked to a disease relevant pathway would be a better course. The resulting methods can be designed and validated rationally. How do you validate quantitative methodology for a nonlinear system when you do not know the structures of hundreds of analytes and you have not paid attention to where the samples came from and what trip they took along the way to your laboratory?


Figure 5: An automated pharmacology system configured for four rodents dosed and sampled asynchronously by a programmed protocol. (Photo courtesy of Bioanalytical Systems, Inc.)
Many faculty members tell us "I'm lucky to get real clinical samples at all. I have no control over their source. We have all sorts of problems with HIPPA and Institutional Review Boards (IRBs) for human subjects." We remind our colleagues of the old data processing adage, "garbage in, garbage out." Our normal mantra is inspired by what we learned from rats. We ask a series of questions. Were the subjects fasted? Do you know their medications? What anticoagulant was used?
How long were the samples in transit from 37 °C to -80 °C? Was this consistent from sample to sample? When were the samples collected? At 8:00 a.m. or 3:00 p.m.? Were some subjects smokers? Which individuals had ham and eggs for breakfast with coffee and which individuals had cereal with herbal tea? You never thought these things would matter? How would you know that if you have never designed an experiment to take into account the influence of diet, diurnal variations, anticoagulants and all the rest?
There are now kits on the market to remove major proteins from plasma before exploring proteomics. Do we know if it makes a difference which of these kits we employ? It makes no difference to the response we see for 500 analytes? Could some key analytes be bound to the major proteins just removed? We would be far more impressed by more thorough validation studies on far fewer samples before a longitudinal study lasting a decade and costing perhaps $20 million was undertaken with no control of factors unknown to make a difference. Would not a month or two of validation work up front be a wise investment?
If we are to look at subtle networks of molecules, be they proteins, peptides, lipids or small molecules, we need to be prepared to look at small changes in the weeds. We are less likely to find factors of five or 10 between health and disease as we once did for some biomarkers that became obvious for this very reason. Glucose and uric acid come to mind.
We do not like the word can't. Funding agencies and the associated reviewers need to pay more attention to these issues and provide the funding for the technician support to get the appropriate data. It is not at all hard, for example, to have subjects in a clinic overnight where diet and sampling times and custody of samples can be tightly controlled. This is done all the time in drug trials and there are some 3500+ beds in the USA alone devoted just to this.
Great progress has been made in -omics with chromatography, mass spectrometry, and NMR. The informatics advances and instrumentation advances result from very heavy investment over decades.
Let us put these advances to work by spending more time on where the samples come from and the odds of success will no doubt be far greater.
If we are to get numbers, let's get good samples.
References
(1) Bioanalytical Method Validation, Guidance for Industry, U.S. Department of Health and Human Serrvices, Food and Drug Administration, Washington, D.C. (2001).
(2) C.T. Viswanathan, S. Bansal, B. Booth, A.J. DeStafano, M. J. Rose, J. Sailstad, V.P. Shah, J.P. Slelly, P.G. Swann, and R. Weiner, The AAPS Journal 9(1), E30-42 (2007).
(3) www.culex.net
Candice B. Kissinger is with the The Chao Center and the Department of Industrial and Physical Pharmacy at Purdue University and Peter T. Kissinger is with the Department of Chemistry.
Michael E. Swartz
"Validation Viewpoint" Co-Editor Michael E. Swartz is Research Director at Synomics Pharmaceutical Services, Wareham, Massachusetts, and a member of LCGC's editorialadvisory board.


Michael E. Swartz
Ira S. Krull
"Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.


Ira S. Krull
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.













Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.





