Investigation of Transferrin Structure via Novel Electron Capture Dissociation Techniques Using a Hybrid Linear Ion Trap Time-of-Flight Mass Spectrometer
Special Issues
Protein and peptide analysis via tandem mass spectrometry (MS-MS) has resulted in a wealth of information regarding protein identification, structure, and abundance levels over the past 10 years. Techniques such as neutral loss scanning and collision-induced dissociation (CID) have been especially helpful in facilitating the identification of a multitude of previously unknown sites of protein phosphorylation. However, many of the techniques used to obtain this information are labor intensive and work inconsistently. To address this problem, much effort has been put forth to find alternative methods of fragmenting peptides and proteins that are less difficult and applicable to a wide gamut of peptide classes. Examples of recently developed dissociation techniques include infrared multiphoton dissociation (IRMPD) and electron transfer dissociation (ETD). The implementation of these new techniques has widened the spectrum of peptides amenable to tandem mass spectral analysis.
Protein and peptide analysis via tandem mass spectrometry (MS-MS) has resulted in a wealth of information regarding protein identification, structure, and abundance levels over the past 10 years. Techniques such as neutral loss scanning and collision-induced dissociation (CID) have been especially helpful in facilitating the identification of a multitude of previously unknown sites of protein phosphorylation. However, many of the techniques used to obtain this information are labor intensive and work inconsistently. To address this problem, much effort has been put forth to find alternative methods of fragmenting peptides and proteins that are less difficult and applicable to a wide gamut of peptide classes. Examples of recently developed dissociation techniques include infrared multiphoton dissociation (IRMPD) and electron transfer dissociation (ETD). The implementation of these new techniques has widened the spectrum of peptides amenable to tandem mass spectral analysis.
Electron-capture dissociation (ECD) is yet an additional peptide fragmentation technique that is gaining in popularity (1). ECD has a number of advantages over traditional fragmentation techniques such as CID. CID tends to cleave the bond between a posttranslation modification (PTM) and its partner peptide before it cleaves the peptide backbone bonds themselves. This premature loss of a PTM during the dissociation process precludes any ability to obtain structural information regarding that PTM. ECD, conversely, tends to leave the bond between a PTM and its partner peptide intact. Instead, it almost universally cleaves the N-Cα bonds of the peptide backbone (except for the N-terminal side of proline). This distinct dissociation pattern makes ECD an extremely useful technique for characterizing PTMs (2).
In addition, ECD has proven to be effective at fragmenting very large peptides, and even small proteins. Indeed, ECD has been applied successfully to ubiquitin, a 76-amino acid protein with a mass of 8564.47 Da. The experiment resulted in a sequence coverage of 85% at the amino acid level (65 of the 76 possible fragments were observed) (3).
While ECD has been an effective method for peptide fragmentation for almost 10 years, historically it has been restricted to costly and complex Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometers, limiting its accessibility to a few specialized laboratories. In addition, the ECD reaction on these instruments occurs on a timescale that is too long to be coupled to a liquid chromatographic separation. Rather, samples must be introduced via direct infusion, prohibiting the study of complex mixtures.
Recently, both of these issues have been resolved with the advent of QuECD, an ECD system that can be coupled to a time-of-flight mass analyzer (4). The QuECD cell comprises a radio frequency linear ion trap that is preceded by a stand-alone linear ion trap and succeeded by a time-of-flight mass analyzer (Figure 1). The initial ion trap accumulates ions of a specific mass and injects them into the QuECD cell, where the ECD reaction occurs. The ions are subsequently transported to the TOF analyzer for high resolution and mass accuracy analysis.


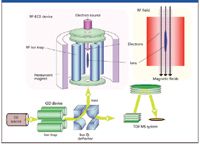
Figure 1: A schematic diagram of the QuECD platform. Specific ions are accumulated and trapped in the initial linear ion trap and directed to the RF-linear ion trap QuECD cell. The resulting fragments are then directed to the time-of-flight tube for high resolution and mass accuracy analysis.
The QuECD system has two prominent advantages over traditional ECD. First, the QuECD reaction occurs on the chromatographic timescale, and is thus capable of analyzing complex mixtures via LC–ECD analysis. The second advantage of the QuECD system is rooted in the fact that the dissociation reaction is achieved by the action of an electron beam focused on the sample within the QuECD cell. Typically, the energy of this electron beam is <1 eV. However, not all peptides fragment efficiently at this energy level. With QuECD, the energy of the electron beam can be modulated to a level that more effectively fragments a particular peptide. "Hot" ECD (HECD) typically is performed at energy levels of 7–10 eV. Some peptides that are not amenable to fragmentation at lower energy levels exhibit very effective fragmentation under HECD conditions.
In this work, we demonstrate examples of both of these extensions to conventional ECD in an analysis of human transferrin. First, we demonstrate QuECD's capability to perform an LC–ECD analysis on a Lys-C transferrin digest. Second, we show the effectiveness of HECD in getting optimal ECD fragmentation of specific transferrin glycopeptides. While these extensions to conventional ECD do not solve all peptide fragmentation problems, they add more tools to the biochemist's arsenal of techniques with which to characterize protein structure, function, and identity.
Experimental Description
Human transferrin (Sigma, St. Louis, Missouri) was resuspended in 8 M urea, 50 mM ammonium bicarbonate, pH 8.5, and subsequently incubated with 10 mM DTT for 60 min at 37 °C, then 20 mM iodoacetamide for 60 min at room temperature. The sample was diluted 10-fold in 50 mM ammonium bicarbonate, pH 8.5, Lys-C was added in a 1:100 Lys-C to protein ratio, and the solution was incubated overnight at 37 °C.


Figure 2: Total ion chromatograms of the (a) LCâCID and (b) LCâECD analyses of a human transferrin Lys-C digest.
The resulting peptides were separated on a 150 mm × 0.05 mm MonoCap C18 column (GL Sciences, Tokyo, Japan) using a 2–35% acetonitrile gradient over 120 min. Figure 2 shows total ion chromatograms for both a traditional LC–MS experiment (utilizing standard CID) and for an LC–ECD experiment. The similarity in the traces indicates that the fundamental change in the tandem MS mode does not alter the basic ability of the instrument to analyze peptides as they are eluted from the column. Of more interest is the similarity between the base peak intensities of the MS-MS events during the chromatographic run (Figure 3). This similarity attests to the ability of QuECD to work on the chromatographic timescale. In fact, the scan speed observed during ECD analysis is only marginally slower than that observed during traditional CID analysis. Over the course of the 120-min LC–MS run, 2340 MS-MS spectra were taken in CID mode, while 1896 ECD MS-MS spectra were recorded, a difference of only 25%. These data demonstrate not only that ECD data can be taken during an LC–MS run, but also that the amount of data generated almost matches traditional CID.

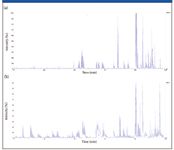
Figure 3: Base peak ion chromatograms of the MS-MS spectra obtained during (a) LCâCID and (b) LCâECD analyses of a human transferrin Lys-C digest.
There are three N-linked glycosylation sites in human transferrin — Asn-432, 491, and 630 (5). We observed ions corresponding to the glycopeptides AA421-433 (3683 Da), AA490-508 (4465 Da), and AA619-646 (5500 Da) during a standard LC–MS experiment. However, we were unable to generate MS-MS spectra for these peptides using traditional CID. In the LC–ECD experiment, however, we were able to obtain ECD spectra for two of these glycopeptides, AA421-433 (m/z 921.5, 4+) and AA619-646 (m/z 1111.0, 4+) (Figures 4a and 4b, respectively). By combining the CID and ECD results, the sequence coverage was increased from 57% to 63%. More importantly, we were able to confirm the location of the glycosylation sites for these two peptides.


Figure 4: ECD spectra of N-glycopeptides of transferrin: (a) AA421-433, 3683 Da, m/z = 921.5 (4+); (b) AA619-646, 5550 Da, m/z = 1111.0 (5+).
Although the ability to perform ECD during an LC–MS run is advantageous, not all peptides have proven amenable to ECD fragmentation. One of the advantages provided by the QuECD platform is the ability to change the energy of the electron beam utilized in the fragmentation process. While traditional ECD reactions are performed at energies of < 1 eV, in HECD, the energy of the electron beam is increased to a value of 7–10 eV. Figure 5 shows ECD spectra of the AA619-646 peptide taken under regular ECD (Figure 5a) and HECD (Figure 5b) conditions. For both the c- and z-ion series, the number of observed ions is markedly increased under HECD conditions. While this ion was not identified using the traditional ECD data, it was easily identified using the richer HECD data.


Figure 5: ECD spectra of transferrin AA619-646 glycopeptide: (a) traditional ECD and (b) HECD.
Conclusion
Modern biochemists require as many tools as possible in the effort to characterize protein structure and function. While MS has long been useful in these efforts, such experiments have proven costly and difficult. Advances in dissociation techniques have made progress in addressing these shortcomings. ECD is a powerful technique because of its utility in characterizing posttranslational modification as well as long peptides and small proteins. Because it can be implemented on a time-of-flight mass spectrometer, the QuECD platform has been successful in increasing the accessibility of ECD to nonspecialized laboratories. Further, QuECD allows for the analysis of complex mixtures via LC–ECD experiments. HECD increases the population of peptides that are amenable to ECD analysis. These advantages have brought QuECD to the forefront of structural protein characterization via MS.
Acknowledgment
The authors would like to thank Takeshi Sakamoto of the Central Research Laboratory, Hitachi Ltd., for his help.
M. Alexander Shaw, Akira Tsuboyama, and Chad Ostrander are with Hitachi High-Technologies America, Pleasanton, California. Makoto Hashimoto and Masaki Watanabe are with Hitachi High-Technologies, Hitachinaka, Ibaraki, Japan.
References
(1) R.A. Zubarev, et al., J. Am. Chem. Soc. 120(13), 3265–3266, 1998.
(2) M.A. Shaw, M. Watanabe, T. Nabetani, Y. Hirabayashi, A. Tsuboyama, and C. Ostrander, Curr. Trends Mass Spec., 26–29, July, 2008.
(3) T. Baba, et al., Hitachi High-Technologies Application Note, NF/TN(E)-002, 2007.
(4) T. Baba et al., Anal. Chem. 76(15), 4263–4266, 2004.
(5) Y. Satomi, et al., FEBS Lett. 576, 51–56, 2004.













Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Analyzing Vitamin K1 Levels in Vegetables Eaten by Warfarin Patients Using HPLC UV–vis
April 9th 2025Research conducted by the Universitas Padjadjaran (Sumedang, Indonesia) focused on the measurement of vitamin K1 in various vegetables (specifically lettuce, cabbage, napa cabbage, and spinach) that were ingested by patients using warfarin. High performance liquid chromatography (HPLC) equipped with an ultraviolet detector set at 245 nm was used as the analytical technique.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


