Improving Separation Performance and Detection Capabilities in Liquid Chromatography Using Active Flow Technology: A Review
LCGC Europe
This review will provide a brief introduction to active flow technology (AFT) with a particular focus on improvements in separation performance that can be achieved when using these columns and coupling with liquid chromatography–mass spectrometry (LC–MS).
The development of sub-2-µm particles, core–shell particles, and monolithic columns has improved separation performance in liquid chromatography (LC) over the past 20 years. However, a key limitation that still prevents LC columns from reaching their full potential is the heterogeneity in the flow profile of solute bands as they travel through the chromatographic packing material within the column. Solutes travel through the column in bowl-like migration profiles, and, as a consequence, more separation power is needed to separate closely eluting solute bands. This limitation can be overcome by using a new type of column technology referred to as active flow technology (AFT). These columns use a new type of column end fitting that negates the problems associated with the bowl-like solute profiles caused by heterogeneity in the fluid flow through the column, which improves efficiency, sensitivity, and therefore increases sample throughput. This review will provide a brief introduction to AFT with a particular focus on improvements in separation performance that can be achieved when using these columns and coupling LC with mass spectrometry (MS).
In liquid chromatographic (LC) separations the shape of the solute band has important implications for the overall performance of the separation. Solute bands should ideally resemble thin flat discs, but rarely do, and more often closely resemble partially filled bowls (1–3). Separation of closely eluting bowl-shaped bands requires greater separation power, because resolution is governed by the separation of the entire solute plug - a thin flat disc resembles the leading edge of the bowl-like band. Peak tailing is therefore an important issue.



(PHOTO CREDIT: KIVILCIM PINAR/GETTY IMAGES)
Bowl-like bands arise from a radial variation in mobile-phase velocity within the stationary phase, which can be caused by a number of factors. Perhaps the earliest known cause was a radial variation in the density and permeability of the stationary phase (3–20). In fully-porous slurry packed particle columns, the density of the stationary phase is highest near the wall, lower in the radial centre, and lowest directly next to the wall (3). The inverse is true for the permeability of the stationary phase bed. This density variation enables the mobile phase to travel fastest at the wall, slower in the radial centre, and slowest near the wall. The reason for this density/permeability variation lies within the packing process.
During packing, particles are suspended as a slurry and pumped at high pressure into the cylindrical column housing; once inside, particles experience shear forces against other particles, with particles being pushed towards the wall. At the wall, friction prevents particles from sliding further causing the bed to pack denser near the wall compared to the radial centre. However, at the wall particles cannot align and close pack completely since neither the wall nor the particles can bend to accommodate the other, leaving voids at the wall. Therefore, a radial density variation is produced (3,21). The story is a little different for columns packed with core–shell particles. The surface of these particles is rougher than their fully-porous counterparts, which causes core–shell particles to experience more friction during the packing process. The result is that core–shell particles move less with respect to each other than fully-porous particles (22). This reduces the radial packing density, and, in turn, velocity variation in core–shell columns. Nevertheless, a packing density distribution still exists, especially at the wall. Radial mobile phase velocity variations of 3% have been reported for core–shell columns packed with 2.7-µm particles compared to a variation of up to 5% in a column packed with fully porous 3-µm particles (20).
In terms of the number of theoretical plates (N), this corresponded to a reduction in N by 33% and 40% for the wall region of the core–shell and fully porous columns, respectively, compared to the radial column centre, which is more chromatographically efficient (20).


Click here to view full-size graphic
Radial mobile phase velocity gradients also arise from viscous heating of the mobile phase, which occurs when the mobile phase is pumped through a column at high velocities. As the mobile-phase is pushed through the stationary phase it experiences friction, and therefore heat. Because heat can dissipate from the wall region of the bed faster than the central region, the central region becomes hotter than the wall region. This causes the viscosity of the mobile phase to be higher in the radial centre compared to the wall region. Consequently a radial velocity gradient is produced (23). The effect of viscous heating becomes more substantial as the particle size decreases, and is particularly important for the sub-2-µm particles used for ultrahigh-pressure liquid chromatography (UHPLC) applications. An additional effect of radial temperature gradients is that the retention factor of the radial central portion of the solute band becomes smaller than that within the cooler wall region, which further distorts the band shape (23). The effect of viscous heating on chromatographic efficiency has been reported as a loss of 20% in terms of N for a column with isothermal efficiency of 15,000 plates, when the column wall was kept at 295 K (23). The effects of viscous heating can be compensated somewhat by using core–shell particle columns because the solid core increases their heat conductivity (24), and by using narrow-bore columns (25,26). However, narrow-bore columns suffer larger long-range eddy diffusion, which reduces their performance compared to wider-bore (4.6 mm i.d.) columns (27,28). Furthermore, when using narrow-bore columns it is important to minimize extra-column dead volume.
It is possible to compensate for the loss of separation power that bowl-like bands produce by using end-point detection. The difference between end-point detection and the bulk detection modes commonly supplied with commercial instrumentation is that end-point detection occurs directly at the end of the stationary phase bed, or at the frit in the case of particle-packed columns. To date, end-point detection has been deployed in either electrochemical (16–20) or optical (9,10) modes where the detector can be placed at precisely defined radial locations. In fact, high efficiency end column detection was introduced as early as 1989 when Baur and Wightman (17) used an electrochemical detector to achieve up to 170,000 plates per metre. The benefit of this type of detection mode is that detection could be performed at the column radial centre (the most efficient part of the column bed), but, unfortunately, this technique is not practical for routine analysis and end column detectors are not commercially available.
Another technique that can compensate for radial velocity gradients is known as "infinite diameter chromatography" (7). The underlying principle or goal of infinite diameter chromatography is that only the uniform radial centre of the column bed is used for separation. To accomplish this, an injection needle is embedded within the top portion of the bed. This means that the sample is directly injected into the cross-sectional radial centre of the column inlet. Adjusting the mobile-phase flow rate, with respect to the column length, and the amount of the stationary phase used in the radial central region of the column enables the solute band to migrate through the column without diffusing into the wall region.
Essentially, this would be a wall-less column, hence the name of the technique. Unfortunately, infinite diameter chromatography is not practical for routine analysis because the needle is embedded into the stationary phase bed and this is not compatible with commercial instrumentation.
Active flow technology (AFT) combines the advantages of infinite diameter chromatography and end-point detection with the practicality of conventional chromatography column formats through the use of a novel column end fitting. Unlike conventional column end fittings that have only one inlet or one outlet port, the AFT column has multiple outlets and multiple inlet ports (Figure 1). Housed inside the AFT fitting is an annular frit that consists of an inner circular frit separated from an outer circular frit by an impermeable ring. By combining this frit and the multiport end fittings it is possible to separate the more chromatographically efficient radial central portion of the solute band from the tailing wall region portion.

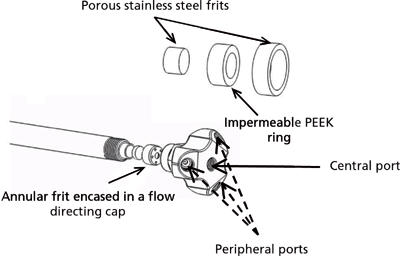
Figure 1: Illustration of an active flow technology column outlet fitting showing the three peripheral exit ports and the single central exit port. The annular frit is expanded and inserts into a cap located within the outlet cap.
The central outlet port on the AFT fitting that extracts the chromatographically most efficient flow from the column can be connected to a typical commercially available detector, and in this mode of operation, an end-point detector is emulated without sacrificing practicality. In this article a brief review will be given of AFT chromatography, covering basic information such as how to operate an AFT column, as well as details on the gains in separation power and sensitivity that active flow technology can provide. The way that AFT can significantly improve the combination of LC with mass spectrometry (MS) will also be discussed. For a more detailed review, the reader is directed to reference (29).
Enhancing Separation Performance Using Active Flow Technology
There are two column formats in AFT: Parallel segmented flow (PSF) and curtain flow (CF). PSF columns have a conventional column inlet fitting and an AFT fitting on the outlet (Figure 1). In PSF, flow segmentation only occurs at the column outlet where the more efficient central part of the solute band elutes from the central port while the less efficient tailing part of the band elutes from the peripheral ports. In CF columns there are two AFT fittings: One on the column inlet, the other on the outlet (Figure 2). Flow segmentation occurs at both the inlet and the outlet in the CF mode. At the inlet of CF columns the autoinjector of the UHPLC or high perfomance liquid chromatography (HPLC) system is connected to the central port while the inlet peripheral ports are connected to another pump, or alternatively, the flow from a single pump can be split pre-injector, such that the appropriate portions of flow are delivered to the central inlet port of the column, via the injector, and the peripheral ports of the column, by-passing the injector. Each mode of operation works equally well, but separate pumping devices provide a more "tunable" system.

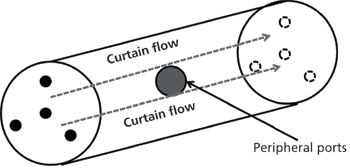
Figure 2: Illustration of the curtain flow chromatography column concept: The column has an AFT fitting on the inlet and the outlet of the column. The inlet central port is connected to the autosampler while another pump or split flow configuration sends mobile phase through the inlet peripheral ports. At the outlet, tubing is used to apply backpressure to adjust the outlet segmentation ratio. Combined, these flow segmentations create a wall of curtain flow that prevents the solute band (in grey) from diffusing into the wall region.
Regardless of the pumping arrangement used, the sample can be loaded into the radial centre of the column and the curtain flow of mobile phase that enters the column through the peripheral port(s) confines the sample to the central region of the column. For example, in preparative CF columns no solute was observed to elute from the peripheral exit ports of the column when 45% of the mobile phase was extracted from the outlet central exit port (Figure 3) (30). It follows that the ratio of flow eluting from the radial central port relative to the peripheral port is an important parameter when operating these columns. This ratio is referred to as the segmentation ratio and is typically reported as the percentage of flow eluting from the central port relative to the total flow eluting from the column.

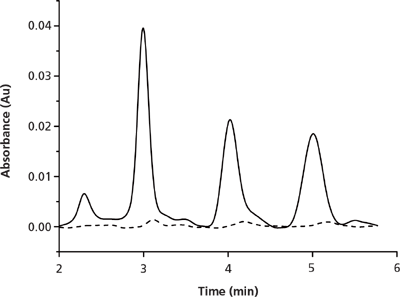
Figure 3: Chromatograms of the eluent from the central port (solid line) compared to the peripheral ports (dashed line) for the preparative scale separation of toluene, propylbenzene, and butylbenzene (listed in order of elution). The inlet segmentation ratio was 22% through the centre. At the outlet a segmentation ratio of 45% through the centre was used. Adapted and reproduced with permission from J. Sep. Sci. 35, M. Camenzuli, H.J. Ritchie, James R. Ladine, and R.A Shalliker, Active flow management in preparative chromatographic separations: A preliminary investigation into enhanced separation using a curtain flow inlet fitting and segmented flow outlet fitting, 410-415 (2012). © John Wiley and Sons.
The segmentation ratio can be changed by controlling the relative pressure drop across the central and peripheral exit ports; in this instance, by using different lengths or diameters of tubing post-detector. For both PSF and CF columns, efficiency in terms of N, sensitivity, and peak height are related to the segmentation ratio. The optimum segmentation ratio depends on the analytical objectives, and cannot be simply stated as a number value. For example, the requirements of a mass spectral detector are different to those of a light attenuating detector, hence there are different optimal segmentation ratios. However, we can state that the sensitivity when using a UV detector located at the radial central exit port where the segmentation ratio is 15% (85% of the flow does not go through the detector) is the same as the sensitivity obtained on a conventional column when 100% of the flow passes through the detector; thus the significance of removing the tailing portion of the solute band.
The efficiency of the PSF column is also dependent on the segmentation ratio and on the retention factor of the solute. This is in part dependent on the management of the post column extra-column dead volume. Generally, the optimal segmentation ratios, with respect to maximizing N, lies in the range between 30% to 45% of the flow through the column centre, but this depends on the column internal diameter.
For example, on narrow-bore columns (2.1 mm i.d.) much higher efficiency is obtained with a segmentation ratio of 20% rather than 43% (data not yet published). The inlet segmentation ratio is more restrictive: Ideally flow-through the central inlet port is around 40% on particle-packed columns, but this is less restrictive on monoliths (31–33). It has been reported that using segmentation ratios of this order for a 4.6 mm i.d. column, on well-managed UHPLC systems operated at high flow rates (4 mL/min) under isocratic elution conditions, gains in the intrinsic efficiency between 90–100% and 70–90% (depending on retention factor) for CF and PSF, respectively (34). The improved efficiency of AFT columns lies in their ability to reduce the effects of trans-column eddy diffusion within particle-packed columns. In short columns (30 × 4.6 mm) packed with 3-µm fully-porous particles, trans-column eddy diffusion can account for 85% of the height equivalent to a theoretical plate (HETP). When AFT columns are used, trans-column eddy diffusion can be reduced by approximately twofold (35).
Sensitivity is also improved with AFT: Gains in sensitivity of almost threefold have been demonstrated for CF columns compared to conventional columns of the same length and diameter (32–34). Performance of AFT diminishes with increasing column length, with the optimal column length being near 150 mm (36).
AFT has also been shown to improve the separation performance of first-generation silica monolithic columns. These columns also suffer the detrimental effects of radial mobile phase velocity variations. However, unlike particle-packed columns, first-generation monolithic columns have a faster mobile phase velocity near the wall, which is more permeable, and a slower velocity in the radial centre of the bed. This has been attributed as a consequence of the sol-gel process used to synthesize these columns (19). When equipped in PSF mode, gains of up to 115% in efficiency and 15% in sensitivity were observed compared to conventional monolithic columns of the same length and diameter (37). Permeability was a big advantage of the first-generation monoliths for high flow rate applications such as multidimensional liquid chromatography. By combining first generation monoliths with AFT, it is now possible to unite the advantage of high permeability with improved separation performance.

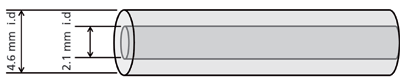
Figure 4: Illustration of the concept of a 4.6 mm i.d. AFT column acting as a virtual 2.1 mm i.d. column.
Using Virtual Columns to Improve the Combination of Liquid Chromatography with Mass Spectrometry
In the previous section, AFT columns were operated at their optimum segmentation ratio to obtain maximum separation efficiency. However, the segmentation ratio can be set so that the wider bore (4.6 mm i.d.) AFT column mimics a narrow-bore column (2.1 mm i.d.). That is, the 4.6 mm i.d. AFT column can act as a virtual 2.1 mm i.d. column housed within a 4.6 mm i.d. column (Figure 4). Using AFT columns as virtual narrow-bore columns has several advantages. Firstly, gains in N by up to 70% and 42% were seen when an AFT 4.6 mm i.d. column was operated as a virtual 2.1 mm i.d. and 3.0 mm i.d. column, respectively, compared to equivalent conventional columns with physical internal diameters the same as the virtual diameters (Figure 5) (38). Secondly, when compared to conventional columns of equivalent diameter, sensitivity in terms of peak height increased by up to 36% and 19% for the virtual 2.1 mm i.d. and 3.0 mm i.d. columns, respectively (Figure 6) (38). These improvements partly arise from the wider bore columns having a greater tolerance for extra-column volume and they are generally more efficiently packed, since the wall surface to column cross-sectional surface area is smaller on the analytical-scale column compared to the narrow bore scale. This was shown by evaluating the efficiency of columns with various internal diameters that were packed with the same stationary phase; 4.6 mm i.d. columns were more efficient (up to 42%) than 2.1 mm i.d. columns, even when the extra-column volume of the system was minimized (28). This was attributed to the smaller trans-column eddy diffusion in 4.6 mm i.d. compared to 2.1 mm i.d. columns (28).

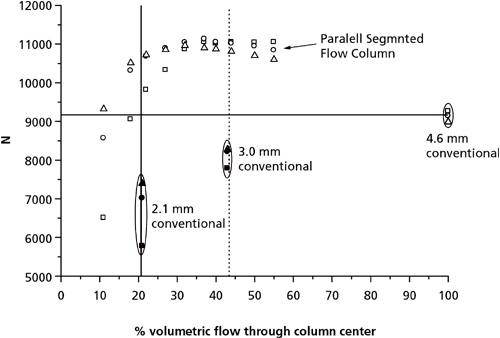
Figure 5: Comparison in N values obtained with a 4.6 mm i.d. PSF column, a 4.6 mm i.d. conventional column, a 3.0 mm i.d. conventional column, and a 2.1 mm i.d. conventional column. Test solutes: Toluene (squares), propylbenzene (circles), and butylbenzene (diamonds). Note the data obtained on the 2.1 mm and 3.0 mm i.d. columns are centred on the 21% and 43% volumetric flow positions to correspond to the equivalent flow through the 4.6 mm i.d. column at that specific segmentation ratio. Adapted and reproduced with permission from Journal of Chromatography A 1262, R.A Shalliker, M. Camenzuli, L. Pereira, and H.J. Ritchie, Parallel segmented flow chromatography columns: Conventional analytical scale column formats presenting as a "virtual" narrow bore column, 64-69 (2012). © Elsevier.
Despite the advantages of analytical-scale columns, LC–MS users prefer to use narrow-bore columns because of the reduced solvent load to the mass spectrometer. This is important because at flow rates above 1 mL/min MS suffers because it relies on the nebulization and evaporation of solvent to produce ions in the ion source. It follows that reducing the solvent load to the mass spectrometer is also important when highly aqueous mobile phases are used. However, separation performance is lost when narrow-bore columns are used. By operating AFT columns as virtual narrow-bore columns, the extra-column dead volume tolerance and improved performance of the wider-bore column format is achieved whilst keeping the solvent load to the mass spectrometer low (38). Recently Kocic et al. (39) coupled a CF column acting as a virtual 2.1 mm i.d. column with MS detection. This allowed chromatographic separations to take place at 5 mL/min without compromising the MS, providing high through-put and greater loadability compared to the narrow-bore column. In fact, sensitivity, in terms of signal-to-noise, improved by 66-fold for some of the amino acids tested compared to the conventional column.

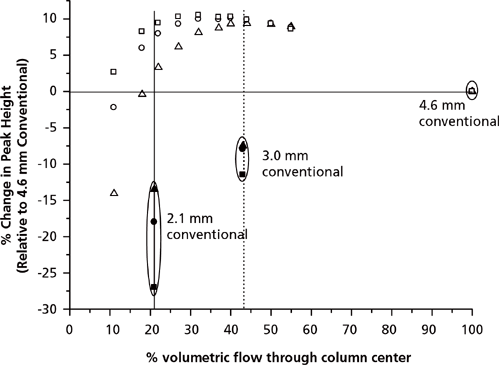
Figure 6: Comparison in sensitivity relative to the 4.6 mm i.d. column. Sensitivity tested on the 4.6 mm i.d. column, the 4.6 mm parallel segmented flow column at various segmentation ratios, the 3.0 mm i.d. conventional column, and the 2.1 mm i.d. conventional column. Test solutes: Toluene (squares), propylbenzene (circles), and butylbenzene (diamonds). Note the data obtained on the 2.1 mm and 3.0 mm i.d. columns are centred on the 21% and 43% volumetric flow positions to correspond to the equivalent flow through the 4.6 mm i.d. column at that specific segmentation ratio. Adapted and reproduced with permission from Journal of Chromatography A 1262, R.A Shalliker, M. Camenzuli, L. Pereira, and H.J. Ritchie, Parallel segmented flow chromatography columns: Conventional analytical scale column formats presenting as a "virtual" narrow bore column, 64-69 (2012). © Elsevier.
A common alternative to using narrow-bore columns with MS is to employ a post-column split with a conventional analytical scale column. Intuitively one may think that using a post-column split would achieve similar results to PSF since both processes split the flow prior to the MS. The performance of a post-column split of 40% was compared to a PSF column with a segmentation ratio of 40% for an LC–MS analysis of 11 amino acids (40). At this segmentation ratio, the PSF column would have dimensions similar to a 3.0 mm i.d. virtual column. The resulting chromatograms shown in Figure 7 (40) clearly show the improved performance of the PSF column compared to the conventional column with a post-column split. Using PSF improved both the sensitivity and the resolution of the separation. This is a direct result of the advantages of the design of the AFT fitting. In PSF, the most concentrated part of the solute profile is sent to the detector while the more diffuse wall region part of the profile is not detected, therefore increasing sensitivity. However, flow segmentation using the post-column split samples the entire band, including the dilute tailing portion. The consequence of this is dilution of the solute as the more concentrated and least concentrated parts of the solute band are combined prior to splitting.

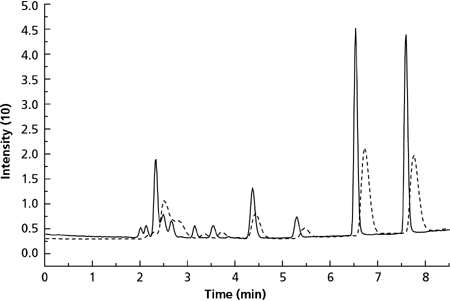
Figure 7: Separation of proline, arginine, cystine, valine, methionine, tyrosine, leucine, phenylalanine, and tryptophan (listed in order of elution). Solid line: Parallel segmented flow (40% through column centre) at 0.5 mL/min total flow through column. Dashed line: Post-column split (40% to MS) at 0.5 mL/min through column. Adapted and reproduced with permission from Rapid Communications in Mass Spectrometry 26, M. Camenzuli, T.A. Goodie, D.N. Bassanese, P.S. Francis, N.W. Barnett, H. Ritchie, J. LaDine, R.A. Shalliker, and X.A. Conlan, The use of parallel segmented outlet flow columns for enhanced mass spectral sensitivity at high chromatographic flow rates, 943-949 (2012). © John Wiley and Sons.
Conclusion
The term active flow technology refers to a new group of LC columns and techniques that enhance the separation power of LC and improves the coupling of LC to volume-limited detectors such as MS. The source of these benefits is the result of a new design of column end fitting, which consists of an annular frit and a multiport header. The purpose of this end fitting is to separate the more efficient, central part of the solute band from the less efficient, tailing part of the solute band, thereby increasing the chromatographic efficiency, sensitivity, and resolution. This brief review focused on two AFT column formats: parallel segmented flow and curtain flow. Not discussed here were the two spin-off advantages of parallel segmented flow - one allows for multiplexed detection, and the other enables highly efficient post-column derivatizations known as reaction flow chromatography. Readers interested in these techniques are referred to references (41–44) for multiplexed detection and (45) for reaction flow.
Michelle Camenzuli is a tenure-track assistant professor within the analytical chemistry group at the University of Amsterdam, The Netherlands. At the moment her research is primarily focused on developing new methods and column technology for two-dimensional liquid chromatography. In 2014 she completed a one year post-doctorate focused on orthogonality in two-dimensional liquid chromatography with Prof. Peter Schoenmakers at the University of Amsterdam. During her post-doctorate she developed a new metric for orthogonality known as the asterisk equations. In 2013 she completed her PhD thesis on the development of active flow technology within the group of Prof. Andrew Shalliker at the University of Western Sydney, Australia. She obtained her MSc majoring in analytical and atmospheric chemistry at Macquarie University (Sydney, Australia) with Dr. Ian Jamie where she studied the effect of rising temperature and carbon dioxide levels on the emission of volatile organic compounds from Australian native trees. She was also the sole analytical chemist at Dyesol Australia and she developed many of their chromatographic separation methods. Dr. Camenzuli currently has 15 publications in peer-reviewed journals and one patent for reaction flow chromatography.
Andrew Shalliker is a professor in analytical chemistry at the University of Western Sydney. He graduated from Deakin University with a BSc (Hons) (1987), a PhD (1992), and a DSc (2012). He has been employed in numerous positions, nationally and internationally. His first appointment was at David Bull Laboratories, followed by a small pharmaceutical company in Mulgrave Victoria (1992–1993). He was an associate lecturer in analytical chemistry at the University of Tasmania (Hobart) (1994). In 1995 he was awarded a QUT Postdoctoral Fellowship (1995–1997). He was a research fellow at the University of Tennessee, Knoxville, USA, and Oak Ridge National Laboratories, Oak Ridge in Tennessee (1997–1999). In 1999 he returned to Australia after accepting a lectureship position at the University of Western Sydney. He now heads a small research group that explores a variety of aspects associated with liquid chromatography. In particular, the development of column technology, stationary phase design, and the separation of complex samples using multidimensional approaches.
References
(1) B.S. Broyles, R.A. Shalliker, and G. Guiochon, J. Chromatogr. A867, 71–92 (2000).
(2) R.A. Shalliker, B.S. Broyles, and G. Guiochon, Anal. Chem. 72, 323–32 (2000).
(3) R.A. Shalliker, B.S. Broyles, and G. Guiochon, J. Chromatogr. A888, 1–12 (2000).
(4) C.E. Schwartz and J.M. Smith, Ind. Eng. Chem. 45, 1209–1218 (1953).
(5) D.S. Horne, J.H. Knox, and L. McLaren, Sep. Sci.1, 531–554 (1966).
(6) S.T. Sie and W.A. Rijnders, Anal. Chim. Acta38, 3–16 (1967).
(7) J.H. Knox, G.R. Laird, and P.A. Raven, J. Chromatogr. 122, 129–145 (1976).
(8) T. Yun and G. Guiochon, J. Chromatogr. A672, 1–10 (1994).
(9) T. Farkas, M.J. Sepaniak, and G. Guiochon, J. Chromatogr. A740, 169–181 (1996).
(10) T. Farkas and G. Guiochon, Anal. Chem. 69, 4592–4600 (1997).
(11) T. Farkas, M.J. Sepaniak, and G. Guiochon, AIChE J. 43, 1964–1974 (1997).
(12) T. Yun and G. Guiochon, J. Chromatogr. A760, 17–24 (1997).
(13) M.S. Smith and G. Guiochon, J. Chromatogr. A827, 241–257 (1998).
(14) K. Miyabe and G. Guiochon, J. Chromatogr. A830, 29–39 (1999).
(15) V. Wong, R.A. Shalliker, and G. Guiochon, Anal. Chem.76, 2601–2608 (2004).
(16) J.E. Baur, E.W. Kristensen, and R.M. Wightman, Anal. Chem.60, 2334–2338 (1988).
(17) J.E. Baur and R.M. Wightman, J. Chromatogr.482, 65–73 (1989).
(18) T. Farkas, J.Q. Chambers, and G. Guiochon, Journal of Chromatography A679, 231–245 (1994).
(19) K.S. Mriziq, J.A. Abia, Y. Lee, and G. Guiochon, J. Chromatogr. A1193, 97–103 (2008).
(20) J.A. Abia, K.S. Mriziq, and G. Guiochon, J. Chromatogr. A1216, 3185–91 (2009).
(21) G. Guiochon, T. Farkas, H. Guan-Sajonz, J.H. Koh, M. Sarker, B.J. Stanley, and T. Yun, J. Chromatogr. A, 762, 83–8 (1997).
(22) F. Gritti, T. Farkas, J. Heng, and G. Guiochon, J. Chromatogr. A1218, 8209–21 (2011).
(23) K. Kaczmarski, F. Gritti, and G. Guiochon, J. Chromatogr. A1177, 92–104 (2008).
(24) F. Gritti and G. Guiochon, J. Chromatogr. A, 1221, 2–40 (2012).
(25) K.D. Patel, A.D. Jerkovich, J.C. Link, and J.W. Jorgenson, Anal. Chem.76, 5777–86 (2004).
(26) M. Martin and G. Guiochon, J. Chromatogr. A1090, 16–38 (2005).
(27) F. Gritti and G. Guiochon, J. Chromatogr. A1333, 60–69 (2014).
(28) F. Gritti and G. Guiochon, J. Chromatogr. A1218, 1592–602 (2011).
(29) R.A. Shalliker and H.J. Ritchie, J. Chromatogr. A1335C, 122–135 (2014).
(30) M. Camenzuli, H.J. Ritchie, J.R. Ladine, and R.A. Shalliker, J. Sep. Sci., 35, 410–415 (2012).
(31) M. Camenzuli, H.J. Ritchie, J.R. Ladine, and R.A. Shalliker, J. Chromatogr. A1232, 47–51 (2012).
(32) M. Camenzuli, H.J. Ritchie, J.R. Ladine, and R.A. Shalliker, Analyst136, 5127–30 (2011).
(33) A. Soliven, D. Foley, L. Pereira, G.R. Dennis, R.A. Shalliker, K. Cabrera, H. Ritchie, and T. Edge, J. Chromatogr. A (accepted manuscript).
(34) F. Gritti and G. Guiochon, J. Chromatogr. A1297, 64–76 (2013).
(35) D. Foley, L. Pereira, M. Camenzuli, T. Edge, H. Ritchie, and R.A. Shalliker, Microchem. J. 110, 127–132 (2013).
(36) F. Gritti, J. Pynt, A. Soliven, G.R. Dennis, R.A. Shalliker, and G. Guiochon, J. Chromatogr. A1333, 32–44 (2014).
(37) A. Soliven, D. Foley, L. Pereira, G.R. Dennis, R.A. Shalliker, K. Cabrera, H. Ritchie, and T. Edge, J. Chromatogr. A1334, 16–9 (2014).
(38) R.A. Shalliker, M. Camenzuli, L. Pereira, and H.J. Ritchie, J. Chromatogr. A1262, 64–9 (2012).
(39) D. Kocic, L. Pereira, D. Foley, T. Edge, J.A. Mosely, H. Ritchie, X.A. Conlan, and R.A. Shalliker, J. Chromatogr. A1305, 102–108 (2013).
(40) M. Camenzuli, T. A Goodie, D.N. Bassanese, P.S. Francis, N.W. Barnett, H. Ritchie, J. LaDine, R.A. Shalliker, and X.A. Conlan, Rapid Commun. Mass Spectrom. 26, 943–949 (2012).
(41) M. Camenzuli, J.M. Terry, R.A. Shalliker, X.A. Conlan, N.W. Barnett, and P.S. Francis, Anal. Chim. Acta803, 154–159 (2013).
(42) M. Camenzuli, H.J. Ritchie, G.R. Dennis, and R.A. Shalliker, Microchem. J., 110, 726–730 (2013).
(43) M. Camenzuli, H.J. Ritchie, and R.A. Shalliker, Microchem. J.110 473–479 (2013).
(44) J.M. Terry, Z.M. Smith, J.J. Learey, R.A. Shalliker, N.W. Barnett, and P.S. Francis, Talanta116, 619–625 (2013).
(45) M. Camenzuli, H.J. Ritchie, G.R. Dennis, and R.A. Shalliker, J. Chromatogr. A1303, 62–65 (2013).























Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

