How to Restore an Idle GC Column to Operating Condition
LCGC Europe
Here is what to do to bring an idle capillary GC inlet column and detector back to operating condition – including column installation, electronic pneumatic control calibration, system bakeout and basic test mix performance.
At some point, you may need to bring an idle capillary GC inlet, column and detector back to operating condition. Here is what to do — including column installation, electronic pneumatic control calibration, system bakeout and basic test mix performance.
A few months ago I needed to use a split–splitless inlet, column and flame ionization detection (FID) system that had been idle for more than a year. The gas chromatography (GC) system had been in continuous use for valved applications, but the inlet carrier gas controller had been disconnected from the carrier gas supply the entire time, and the column oven connections and gas inlets had been capped. The intended column — 20 m × 180 µm, 0.12-µm df methylsilicone fused silica — had been stored in its box with the inlet and outlet stuck into a septum. The GC instrument itself was apparently in excellent working condition, so I optimistically anticipated a quick install and performance checkout. Fortunately, that prediction proved accurate. Here are the steps that I followed.
Instrument Setup
My initial task was to set up the pneumatics, inlet and detector. First, with the GC system powered on, I checked the carrier gas configuration for the split–splitless inlet. Although the set column dimensions did not match the column I would use, the basic configuration would allow carrier to flow to the inlet once a supply was connected. I also made sure that carrier gas leak detection was disabled. I planned to purge the pneumatics initially and did not want the system to shut off the flow. I then doublechecked that the inlet and FID heaters were turned off and also turned off the detector and pneumatic controllers that had been in use.
Pneumatics
Next, I turned off the GC system power, allowed its zones to cool, removed the existing columns and capped off the detector and carrier gas source that had been in use. I then removed the interconnecting tubing at the gas switching valves while making a note of how they were connected. The new application would require oven temperatures of up to 300 °C, well above the maximum limits for the gas sampling valves, so I disconnected the valves from their actuators and removed them as well.
For my next step, I inspected the gas tanks. I found suitable gas purities: 99.9999% research-grade helium carrier gas, zero grade air with < 1 ppm total hydrocarbons, and highpurity 99.998% hydrogen. The carrier-gas regulator was a stainless steel diaphragm, high-purity dual-stage device and the fuel gas regulators were brass dual-stage types. Moving down the installed copper gas lines, I found a suitable set of multistage gas filters that were in apparent good condition with littletono indicated filter exhaustion.
I turned off the carrier gas at the tank, moved the carrier-gas supply over to the pneumatic controller for the split–splitless inlet system, but did not tighten the connection completely, and capped the carrier-gas connection that had been used for the valved application. While making the carriergas connection, I noted that the existing tubing and ferrules appeared to not have been overtightened or scratched and that the nut screwed onto the fitting smoothly with no binding.
Before fully tightening the new carriergas connection, I turned the gas on at the tank and allowed some helium to purge through the line. This would protect the upstream filters by removing the air that had inevitably diffused in while the tubing was disconnected. Of course, any additional air present in the pneumatic controller would enter the line as well, so after tightening the fitting onequarter turn past finger tight I immediately turned on the GC system and set a split flow of 50 mL/min at a 10-psig (70-kPa) inlet pressure.
I also turned on the air and hydrogen supplies, verified that the regulator output pressures were correct and then enabled the FID fuel gas flows and checked that they were set to the correct values as well, according to the instrument manual.
After I was satisfied that the gas lines were purged sufficiently, I turned off the gas flows at the instrument pneumatic controllers and then closed the tank valves to commence a gross leak check. After 10 min, I turned the tank valves back on one at a time and observed any motion of the highpressure gauges on the pressure regulators. An observable change in the high-pressure gauges that is greater than the wiggle produced by lightly tapping the gauges may indicate a serious leak that requires further investigation. I saw no discernible movement, so I proceeded to do a fine leak check of the hydrogen and helium lines with a leak-check device. On its high-sensitivity setting, the leakchecker probe picked up a small leak at the hydrogen filter fitting, but I was able to seal it completely by tightening the fitting one-eighth turn. Otherwise, I would have chosen to cut the tubing an inch shorter and remake the fitting with a new nut and ferrules instead of tightening the leaking fitting further.
At this point, the instrument had been powered on for the better part of an hour, so it was time to zero the pressure transducers, which need some time to stabilize after powering on. The largest drift error in the pressure transducers is their zero reading. Span errors normally are not corrected, as that would require a very accurate (and expensive) external gauge, and in doing so one would be just as likely to introduce errors as to correct them.
Following the procedure in the manual, I turned off the gas supplies and allowed the line pressures to decay to zero, which took another 10 min or so. Then I accessed the pneumatic zeroing section of the instrument firmware and set the zeros. I finished by turning the gas supplies back on.
Inlet and Detector
I had to assume that someone might have fiddled with the components during the year of idle time, so after turning off the instrument power again I proceeded to check the condition of the inlet and detector. I like to wear lint-free gloves while handling inlet and detector parts, but I've used tweezers or pliers (gently) as well.
Although there was nothing apparently wrong with them, I removed and replaced the inlet septum, liner and inner seals. While the inlet was disassembled I checked the column connection for bits of ferrule and the inlet cap for pieces of septum. Then I reassembled the inlet with new parts.
I removed the detector top, electrode and collector tube from the FID system. They were clean enough so I simply put them back. The upcoming performance check would bring out any problems in that area. I also checked the FID fitting in the oven to make sure the inner passageway was clear. Before the next step, I sealed the FID oven connection with a blank graphite-vespel ferrule.
Then it was time to check the FID fuel gas flow calibration. I attached a digital flowmeter, recently calibrated, to the FID tower outlet and enabled the air flow first. The flow was within a few standard cubic centimetres of the set point, so I turned off the air flow and turned on the hydrogen flow. After remembering to switch the flowmeter to its hydrogen scale, I found the flow to be close to the set point as well. If this instrument had makeup gas flow for the FID system (which it did not), then I also would have checked that flow rate at this point. I made sure that the hydrogen flow was turned off before continuing.
When I have new inner parts in an inlet, I like to bake it out before installing the analytical column. This step will remove much of the inevitable contamination from the inlet. I installed an old 250-µm i.d. column I had on hand for such purposes, turned on the GC system and set the carrier gas to 10 psig and 200 sccm of split flow. I did not connect the column outlet. After verifying that the split vent flow was present and calibrated, I checked around the inlet seals and column fitting for leaks. Then I set the inlet temperature to the maximum application temperature, plus 20–300 °C in this case. The inlet was a programmable temperature device, so it heated up quickly. I set the GC oven to the bakeout temperature of the column to be installed later, also 300 °C, allowed the oven to stay there for 10 min, and then cooled the oven and inlet back down.
Naturally, since this oven had not been heated that high in quite a while, I had to assure some of my coworkers that, no, there was no fire or electrical short — that the burning smell was normal and that I was ok. It was a great way to find out who's watching out for their colleagues.
Column Installation and Checkout
With the inlet baked and cooled, I was ready to install the analytical column. Like most used fused silica GC columns, this one would be somewhat shorter than the originally specified length written on the box. I measured the approximate average coil diameter and counted the number of turns of column on its cage to check the length and get an estimate of how much was left: This came to 35 turns at 17.5-cm diameter, for an estimated length of 19.25 m.
It is important to know the column dimensions because electronic pneumatic control systems calculate pressure, flow and velocity on the assumption that the analyst has entered the correct dimensions. To the extent that the column dimensions are incorrect, the operating conditions also will be erroneous. Large errors, such as operating a 250-µm i.d. column with the pneumatic system set for a 320-µm i.d. column, should become obvious in short order, but smaller discrepancies might not be seen as clearly. I entered the column box dimensions, with a length of 20.0 m, an internal diameter of 180 µm and a film thickness of 0.12 µm, to find out what the effects of incorrect column dimensions would be.
The thin stationary film would not contribute significantly to the pneumatic operation, but it's a good idea to be in the habit of always entering all of the column dimensions whenever installing a column. I also checked that the carrier gas was set to helium and that the split mode was set to flow control, not split ratio mode.
Using new ferrules, I installed the column at the inlet. Keeping the same 10 psig inlet pressure, I then turned on the inlet carrier gas and doublechecked for column flow by putting the column outlet into an autosampler vial that was half-filled with distilled water. The presence of bubbles revealed carrier flow. I then connected the column outlet to the detector. After a moment or two, I leak-checked the column inlet connection. There were no problems there.
I activated the FID system, set the temperature to 300 °C and allowed it to heat until the temperature exceeded 150 °C. Then I turned on the hydrogen and air flows and gave the ignite command through the keypad. In a moment, I heard the short pop of flame ignition. I double-checked for a flame by holding a cold wrench horizontally over the FID exit to observe some water condensation. The FID signal level had risen as well, verifying that the flame was staying lit.
Verifying the Column Dimensions
With the column installed, flame lit and carrier flowing, it was time to verify that the pneumatic controller had column dimensions that were close enough to provide accurate readings of flow and velocity. I set the oven to 120 °C and entered a linear velocity of 40 cm/s. As the oven stabilized, the inlet pressure settled to 29.4 psig (202 kPa) and I was ready to inject some methane.
I chose to use the average linear velocity as my metric because this value is easily measured with sufficient accuracy. Column flow could be used instead, but that would require comparably accurate measurement of low flows, around 1 mL/min for the present column, which is very difficult to achieve even if the column is removed from the detector.
I set up a short, 2-min run on the data handling system and injected 10 µL of 1% methane in nitrogen to capture the unretained peak time and shape. Figure 1 shows the result. The methane peak is symmetrical and the baseline noise is quite low. On closer examination, the noise appeared to be comparable to that found in previous chromatograms on this instrument, so I did not concern myself with it. This result signifies that the system is tight, with no serious detector leaks or poor electrical connections, and that the inlet system and column are free of obvious obstructions or misconfigured connections. A tailing methane peak would indicate a problem in the inlet liner or column connection areas, or possibly blocked or incorrect makeup gas flow at the detector.


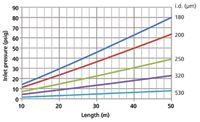
Figure 1
However, the calculated average linear velocity from the 44-s unretained peak time was 45.5 cm/s for the nominal 20-m column length, not the set value of 40 cm/s. Although this approximately 10% error is not huge, it is instructive to consider possible sources for the discrepancy. Errors of this sort will result if the column length or diameter, the oven temperature (as it affects the carrier gas viscosity) or the pressures are not conveyed accurately to the electronic pneumatic system.
Theoretical equations can be used to calculate the range of inlet pressures required to produce an average velocity of 40 cm/s with helium at 120 °C, for example, with varying column lengths and internal diameters. I used a spreadsheet for this, to produce the family of curves illustrated in Figure 2. The curve for a 20 m × 180 µm column passes through our pressure setpoint of 29.4 psig (202 kPa) as expected, yet the measured average velocity was greater than 40 cm/s. A GC pressure–flow calculator application (1) can also be used to examine the effects on flow and velocity of changing column dimensions. Interested chromatographers can read more about the theoretical calculations on an external website (2).

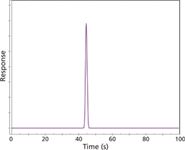
Figure 2
The initial unretained peak measurement and velocity calculation implied that either the inlet pressure drop was greater than its set level, the column was shorter than the entered 20 m, the column internal diameter was slightly larger than the set value of 180 µm or the oven temperature was miscalibrated.
The oven temperature was the least likely culprit. This GC system had been temperature-calibrated in the past three months and an error of 10 °C would only change the viscosity, and thus the velocity, by less than 2% anyway. I wasn't too concerned about the temperature.
I already knew that the column was shorter than its original 20 m: How much shorter would it need to be to achieve the desired 40 cm/s from the electronic pneumatic system? To answer this question, I measured a series of unretained peak times across a range of pressures that I guessed would cover the true 40-cm/s operating point. Since the average velocity is a function of the length as well as the pressure, I used a spreadsheet to calculate the theoretical linear velocities as a function of the pressure and length. The theoretical predictions matched the measured data well when the column length was 18.75 m, as shown in Figure 3(a). When the applied column length diverged much from 18.75 m, then the experimental data could no longer be said to match the theory.


Figure 3
I had measured the column length at 19.25 m. Is a 50-cm error in the length reasonable given how it was measured? With 55 cm/turn, a one-turn counting error would account for it. Or, an error of 0.5 cm measuring the coil diameter would produce a similar effect across the 35 turns. After working out these data, I retrieved the column and carefully remeasured the coil diameter and turns count, twice. However, the results were the same. The coil diameter appeared to be accurate to about 0.15 cm; the turns count was unchanged. Thus, it seemed that the column length alone was insufficient to account for all of the effects on linear velocity.
The column internal diameter could also play a role in the linear velocity discrepancy. Pressure drops are sensitive to the square of the diameter, so a small difference in the actual versus nominal diameters could also be in play. I set the theoretical column length to 19.25 m, and calculated that the inner diameter would need to be around 185 µm to achieve a 40 cm/s average velocity at an inlet pressure of 26.6 psig. Figure 3(b) shows this result. I also noted that if the column length were assumed to be 20 m, the inner diameter would have to be about 188 µm to account for the observations.
Generally, column internal diameters are quite accurate. They are controlled to tight specifications by precision laser gauges during column tubing production. One publication cites the internal diameter of similar tubing as 180 ± 5 µm (3), so an upper limit of 185 µm is not out of reach.
Finally, perhaps the inlet pressure was not calibrated, although it had been zeroed. Would a positive or negative pressure offset account for the measured velocity data? Yes, indeed. A positive offset of +1.75 psia (12 kPa) would be sufficient, as shown in Figure 3(c), to produce the velocity effect in lieu of a larger column internal diameter. Modern pressure control systems should be capable of much more accurate operation, but perhaps this 10-year-old GC system was not up to the challenge.
From the above considerations, the most likely source of error in the matching of linear velocity set point and measurement was the analyst; in this case, myself. It was highly suspicious that exactly one turn of column coil length would account for the results, yet the column length seemed to have been measured accurately. Some combination of pressure, diameter and length could be operating in combination, but simplicity, and the fact that columns are trimmed routinely, led me to conclude that compensating by entering an adjusted column length was the best way to bring linear velocity in line.
Short of uncoiling the column and measuring how many tiles it spans on the laboratory floor — which I did once a long time ago and failed to recover a neatly coiled column in one piece after someone stepped on it — the next best way to measure length is the turns method; just be careful to get an accurate turn count and diameter measurement. Count twice and inject once. Use the measured length as a starting point, then use a GC pressure–flow calculator to figure out an adjusted length to enter into the pneumatic controller so that the velocity readout is reasonably accurate.
If large adjustments seem necessary to align the measured and displayed linear velocities, then question the entire setup and be prepared to review each step of the system configuration and installation process.


Figure 4
Bakeout and Performance Test
Finally, I baked the column out at 300 °C (twice), and then ran a polarity test mixture to gauge its performance. Figures 4 and 5 show these results, which were well within expectations with the exception of the slightly tailing solvent peak. This tailing peak had the look of a minor detector problem typical of chlorinated solvents, such as the methylene chloride in use here, but since the analytes were not affected I chose to ignore it. With a well characterized column up and running, I was ready to proceed to the application itself.

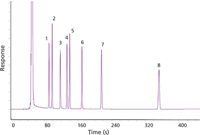
Figure 5
Conclusion
Restoring an idle capillary inlet and detector system can be straightforward if the inlet, column and detector were sealed and stored properly. Conducting fairly simple pneumatic and hardware checkouts and leak checks before attempting to run any chromatograms will hasten the restoration process. Given that few capillary columns come back to the GC oven with the same length, as originally stated on the box, be prepared to spend a little time adjusting electronic pressure control settings if accurate flow and velocity readings are desired.
"GC Connections" editor John V. Hinshaw is senior research scientist at BPL Global Ltd, Hillsboro, Oregon, USA and a member of LCGC Europe's editorial board. Direct correspondence about this column should go to LCGC Europe editor, Alasdair Matheson, 4A Bridgegate Pavilion, Chester Business Park, Wrexham Road, Chester, CH4 9QH, UK or email amatheson@advanstar.com
References
(1) See the Agilent pressure–flow calculator application and software at http://www.chem.agilent.com/en-US/Support/Downloads/Utilities/pressureflowcalc/Pages/default.aspx.
(2) http://wiki.hrgc.com.
(3) S. Griffin, LCGC N. Amer., 20(10), 928–938 (2002).














University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.

Miniaturized GC–MS Method for BVOC Analysis of Spanish Trees
April 16th 2025University of Valladolid scientists used a miniaturized method for analyzing biogenic volatile organic compounds (BVOCs) emitted by tree species, using headspace solid-phase microextraction coupled with gas chromatography and quadrupole time-of-flight mass spectrometry (HS-SPME-GC–QTOF-MS) has been developed.



