Host Cell Protein Monitoring During Downstream Processing Using Micro-Pillar Array Columns Combined with Mass Spectrometry
LCGC Europe


This article investigates host cell protein analysis using micro-pillar array columns combined with mass spectrometry.
Protein biopharmaceuticals have substantially reshaped the pharmaceutical market and today over 350 products have been approved for human use in the United States and the European Union. Protein biopharmaceuticals are commonly produced recombinantly in mammalian, yeast, or bacterial expression systems. Next to the therapeutic protein, these cells produce endogenous host cell proteins (HCPs) that can contaminate the biopharmaceutical product despite multiple purification steps in a process. Since these processârelated impurities can affect product safety and efficacy, they need to be closely monitored. Enzyme-linked immunosorbent assays (ELISA) are recognized as the gold standard for measuring HCPs because of their high sensitivity and high throughput. Mass spectrometry (MS), however, is gaining acceptance as an alternative and complementary technology for HCP characterization. This article reports on the use of micro-pillar array columns combined with MS for the characterization and in-depth monitoring of HCPs during downstream processing.
In contrast to small molecule drugs, which are commonly synthesized by chemical means, protein biopharmaceuticals result from recombinant expression in bacterial, yeast, and mammalian cells. As a consequence, the biotherapeutic is co-expressed with hundreds of host cell proteins (HCPs) with different physicochemical properties present in a wide dynamic concentration range. During downstream processing, the levels of HCPs are substantially reduced to a point considered acceptable to regulatory authorities (typically < 100 ppm – ng HCP/mg product). These process-related impurities are considered as critical quality attributes because they might induce an immune response, cause adjuvant activity, exert a direct biological activity (for example, cytokines) or act on the therapeutic itself (for example, proteases) or excipients (for example, phospholipase on co-formulated Tween) (1,2,3).
Multicomponent enzyme-linked immunosorbent assays (ELISA) is currently the workhorse method for HCP testing because of its high throughput, sensitivity, and selectivity (1). Polyclonal antibodies used in the test are typically generated by the immunization of animals with an appropriate preparation derived from the production cell minus the productâcoding gene. However, ELISA does not comprehensively recognize all HCP species, that is, it cannot detect HCPs to which no antibody was raised, it only provides information on the total amount of HCPs without providing insight into individual HCPs and, in a multicomponent setup, it has poor quantitation power. In that respect, mass spectrometry (MS) nicely complements ELISA because it can provide both qualitative and quantitative information on individual HCPs. In recent years, various papers have appeared dealing with the mass spectrometric analysis of HCPs (4–23). These studies typically rely on bottomâup proteomic approaches in which peptides derived from the protein following proteolytic digestion are handled. Evidently, one is confronted with an enormous complexity and dynamic range and the separation space is dominated by peptides derived from the therapeutic protein. For successful HCP analysis, there is clearly a need for highly efficient up-front MS separations. In that respect, micro-pillar array columns are highly promising. The origin of this technology dates back to the late 1990s when Regnier et al. addressed the problem of miniaturizing capillary electrochromatography (CEC) columns and introduced microfabricated supports as an alternative for the conventional packed beds (24,25). The theoretical benefit (reduction of the van Deemter A-term) of such supports was elucidated only a few years later by Knox (26). In the years to follow, Desmet et al. conducted several quantitative studies on Knox’s argument, taking a column filled with an array of pillars as a representative example (27). Finally, in 2007, the first proof-of-concept of micromachined liquid chromatography (LC) columns operated by pressureâdriven liquid flow, later termed micro-pillar array columns was reported (28). The inherent high permeability and low “on-column” dispersion obtained by the “perfect order” of the separation bed makes micro-pillar array columnâbased chromatography unique and offers several advantages compared to conventional column technologies (packed beds and monoliths). The peak dispersion originating from heterogeneous flow paths in the separation bed is eliminated (no A-term contributions) and therefore components remain much more concentrated (sharp peaks) during separation. The freestanding nature of the pillars also leads to much lower back pressure, which permits the use of very long columns. These properties result in excellent chromatographic performance with high resolution and high sensitivity. This article reports on the use of micro-pillar array columns combined with MS for the characterization of HCPs and their monitoring during downstream processing.
Materials and Methods
Materials: Water and acetonitrile were purchased from Biosolve. Formic acid (FA), trifluoroacetic acid (TFA), dithiothreitol (DTT), and 2-iodoacetamide (IAA) were from Sigma-Aldrich. Tris-HCl pH 7.5 was purchased as a 1 M solution from Thermo Fisher Scientific. Porcine sequencing-grade modified trypsin was acquired from Promega. Rapigest, MassPREP enolase (ENL), bovine serum albumin (BSA), alcohol dehydrogenase (ADH), and phosphorylase b (PHO) digestion standards were purchased from Waters. Downstream process samples were obtained from a local biotechnology company.
Sample Preparation: To a volume corresponding to 100 µg of protein, 105 µL of 0.1% Rapigest in 100 mM Tris-HCl pH 7.5 was added followed by the addition of 100 mM Tris-HCl pH 7.5 to a final volume of 192.5 µL. The sample was subsequently reduced at 60 °C for 30 min by the addition of 5 mM DTT (2.5 µL of 400 mM DTT in 100 mM Tris-HCl) and alkylated at 37 °C for 1 h by adding 10 mM IAA (5 µL of 400 mM IAA in 100 mM TrisâHCl). Digestion proceeded for 16 h at 37 °C using trypsin as protease added at an enzyme to substrate ratio of 1:25 (w/w). Lyophilized trypsin (20 µg) dissolved in 100 mM TrisâHCl (50 µL) was added in a volume of 10 µL giving rise to a final sample volume of 210 µL. Rapigest removal was achieved by adding TFA to a final concentration of 0.5%. Following incubation for 30 min at 37 °C, the sample was centrifuged for 10 min at 13,000 rpm and the supernatant was carefully transferred to an autosampler vial. MassPREP ADH, PHO, BSA, and ENL digestion standards were added at a concentration of 1000 fmol, 100 fmol, 50 fmol, and 10 fmol / 100 µg therapeutic, respectively.
LC–MS: An Ultimate 3000 RSLCnano system (Thermo Scientific) was used for LC–MS measurements. Tryptic digests were analyzed on a 200 cm C18 µPAC column (PharmaFluidics) at 50 °C. Samples were loaded on a 1 cm C18 µPAC trapping column (PharmaFluidics) using 0.1% TFA in 1:99 acetonitrile–H2O (v/v) at a flow rate of 10 µL/min. Elution was carried out with a gradient of (A) 0.1% FA in H2O and (B) 0.1% FA in 80:10 acetonitrile–H2O (v/v) from 1% B to 30% B in the first 115 min and from 30% B to 45%B in the following 11 min. The flow rate was 750 nL/min from 0 to 10 min and 500 nL/min from 10 min to 150 min. An injection program allowed the introduction of 4 µL of sample in between two plugs of loading buffer. Loop size was 20 µL and samples were kept at 10 °C in the autosampler tray while waiting for injection.
High-resolution accurate mass measurements were performed on a Q Exactive HF-X Hybrid QuadrupoleâOrbitrap mass spectrometer (Thermo Scientific) equipped with a Nanospray Flex source (Thermo Scientific). The micro-pillar array column was connected via a 20 µm internal diameter (i.d.)/360 µm outside diameter (o.d.) fused-silica capillary to a PicoTip emitter (10 µm tip i.d. – New Objective) via a 50 µm bore stainless steel union (C360UFS2 360 µm union from VICI AG International) and true no twist one-piece PEEK fittings (C360NFPK 360 µm fittings for fused-silica tubing from VICI AG International). Spray voltage was set at +2.0 kV, the capillary temperature was 275 °C, and a S-lens RF level of 40 was used. The Q Exactive HF-X was operated in DDA (data dependent acquisition) mode, where one cycle consisted of a MS1 survey scan followed by TopN MS/MS scans. MS1 spectra were collected in centroid mode in a scan range from 380 m/z to 1500 m/z at a resolution of 120,000 at m/z 200. Automatic gain control (AGC) parameters included 3e6 target value and maximum injection time of 100 ms. The ten most abundant multiplyâcharged precursors were selected for fragmentation by higherâenergy collisional dissociation (HCD) (28% NCE). MS/MS spectra were acquired at 15,000 resolution at m/z 200 and MS2 AGC parameters included target of 5e4 and maximum injection time of 200 ms. The precursor isolation window was set to 1.2 Th and dynamic exclusion was set to 90 s.
Protein Identification and Quantification: Data files were processed using Proteome Discoverer (v2.3) (Thermo Scientific). Spectra were searched against protein sequences consisting of CHO-K1 protein database (08/24/2014) (downloaded from www.chogenome.org), therapeutic mAb fragment, and ADH, PHO, BSA, and ENL using the Sequest HT search engine. Search parameters were precursor mass tolerance of 10 ppm, fragment mass tolerance of 0.02 Da, carbamidomethylation of cysteine was selected as a fixed modification, and a maximum of two trypsin missed cleavages were allowed. False discovery rate (FDR) targets were set to 0.01 (99%) and 0.05 (95%) as strict and relaxed, respectively, calculated by the target decoy peptide spectral match (PSM) validator node. Keratin and trypsin peptides and proteins were excluded from the final list.
Protein abundances, calculated from the average of the three most abundant unique peptides, were obtained via the Precursor Ions Quantifier node in the Proteome Discoverer software.
Results and Discussion
The separation beds of microâpillar array columns are fabricated by etching the interstitial volumes out of a silicon substrate following lithographic definition of an array of pillars. This creates a stationary phase support structure that is organized in a reproducible and ordered pattern. Concatenation of several of these channels allows long column lengths to be fabricated on a small footprint (29). The most important characteristics of the micro-pillar array separation bed design are: pillar diameter, 5 μm; inter pillar distance, 2.5 μm; pillar height or bed depth, 18 μm; external porosity (Vinterstitial/Vtotal), 59%; bed channel width, 315 μm; and bed length, 200 cm. To increase the retentive surface, the pillars are rendered superficially porous with a typical porous shell thickness of 300 nm and pore sizes in the nanometre range. The porous surface has been uniformly modified with octadecyl chains to create a hydrophobic stationary phase suited for reversedâphase LC separations. Figure 1 shows some relevant characteristics of the micro-pillar array column.



Because of the high permeability, the 200 cm column used in this study can be operated at moderate LC pump pressures (50 to 300 bar) over a wide range of flow rates (100–1000 nL/min). Van Deemter measurements with heptyl-phenyl ketone demonstrated that a total of 400,000 theoretical plates could be generated at the optimal linear solvent velocity, corresponding to a flow rate of 200–250 nL/min and generating a column back pressure of only 70 bar.
In this study, micro-pillar array columns were used in combination with a hybrid quadrupole-orbital trap MS system for the characterization of HCPs throughout the downstream processing of a therapeutic mAb fragment recombinantly expressed in Chinese Hamster Ovary (CHO) cells. Proteinaceous samples collected at different purification steps were reduced using DTT and alkylated using IAA prior to overnight trypsin digestion. Two µg of digested sample, spiked with four pre-digested protein calibrants (ADH at 10 fmol/µg, PHO at 1 fmol/µg, BSA at 0.5 fmol/µg, and ENL at 0.1 fmol/µg), was subsequently loaded (via a pillarâbased pre-column) onto a 200 cm long micro-pillar array column. Peptides were separated using a 116 min gradient and the MS system was operated in DDA mode. Data were subsequently searched against the CHO database extended with the sequence of the mAb fragment and the four protein calibrants (ADH, PHO, BSA, and ENL).


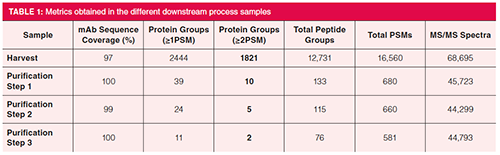
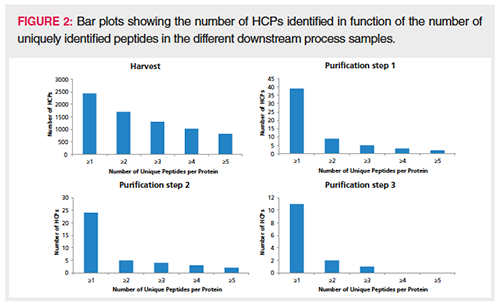
Table 1 shows some important metrics obtained on the downstream process samples. In the harvest sample, close to 70,000 MS/MS spectra were obtained, and, following database searching, 12,731 unique peptides could be identified corresponding to 2444 protein groups with at least one PSM and 1812 proteins with more than one PSM. A nice clearance of these proteins throughout the purification process was observed and in the last purification step the number of HCPs could be reduced to two. Note that in the table, the mAb fragment, ADH, PHO, BSA, ENL, as well as keratin and trypsin, are excluded from the protein groups columns. Figure 2 plots the number of HCP protein identifications by function of the number of uniquely identified peptides. In the harvest sample, a substantial number of HCPs are identified with more than one peptide. In the downstream purification samples, one-hit wonders start to dominate as a result of the much lower abundance of HCPs.


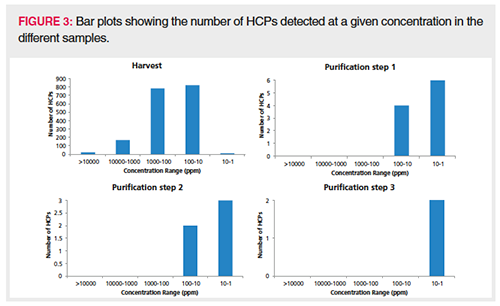
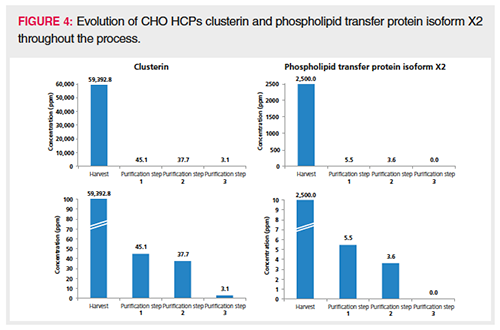
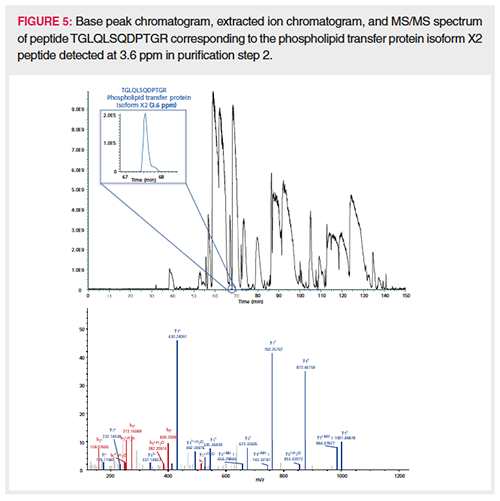
Semiquantitation of the HCPs identified with more than one PSM was achieved by spiking pre-digested proteins (ADH, PHO, BSA, and ENL) at known concentrations in every sample. The average intensity of the three most intense peptides for each protein calibrant was plotted in function of column load (expressed in fmole), and the slope of the resulting calibration curve (counts/fmole) was applied for semiquantitation of the HCPs in the corresponding sample. This is an adaptation of the procedure described by Silva et al. (30) and commonly used in HCP quantitation. The average slope measured in the different downstream samples was 15,918,593 counts/fmole with a relative standard deviation (RSD) of 14.1%. This precision allows for a good comparison in HCP quantity throughout the process. Upon backâcalculating the concentration of the spiked protein digests (ADH, PHO, BSA, ENL) in every sample using the corresponding slope, an accuracy well between 50% and 200% was obtained, which is more than acceptable for semiquantitation. Figure 3 plots the number of HCPs detected at a given concentration in the different samples. The bar plot presented in Figure 4 follows two specific HCPs throughout the process. Clusterin, for example, is present at 59,393 ppm in the harvest but is reduced to 3.1 ppm in the final purification step. Clusterin can easily associate in a nonspecific manner with both the Fc and Fab fragments of antibodies (2,31). Phospholipid transfer protein isoform X2 is present at 2500 ppm in the harvest and reaches 0 ppm in the final purification step. Figure 5 shows the extracted ion chromatogram of one of the phospholipid transfer protein isoform X2 peptides detected at 3.6 ppm overlaid with the base peak chromatogram. This figure illustrates the large protein dynamic range within the sample, one of the major challenges in HCP characterization. This particular HCP was present at low ppm levels, however, high-quality MS/MS data could be obtained allowing its confident identification. When surveying the base peak chromatogram, it becomes clear that the column is overloaded at 2 µg. The broad peaks observed can all be traced back to the therapeutic antibody. Peptides originating from the HCPs on the other hand give nice Gaussian and sharp peaks.






Conclusion
The method described is useful for the characterization of HCPs and their monitoring throughout downstream processing. Downstream process samples were directly digested following reduction and alkylation and the resulting peptides separated using one-dimensional (1D) LC. The MS system was operated in data dependent mode. HCP content and sensitivity can potentially be further improved by implementing a second chromatographic dimension (two-dimensional [2D]-LC) (5,12), by optimizing sample preparation by depletion or HCP-directed digestion (14,17), by MS-based exclusion of precursors originating from the therapeutic itself, or by operating the MS system in data independent mode (17).
References
- F. Wang, D. Richardson, and M. Shameem, BioPharm Int.28, 32–38 (2015).
- M. Vanderlaan, J. Zhu-Shimoni, S. Lin, F. Gunawan, T. Waerner, and K.E. Van Cott, Biotechnol. Prog.34, 828–837 (2018).
- S.X. Gao, Y. Zhang, K. Stansberry-Perkins, A. Buko, S. Bai, V. Nguyen, and M.L. Brader, Biotechnol. Bioeng. 108, 977–982 (2011).
- M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Anal. Biochem.428, 150–157 (2012).
- C.E. Doneanu, A. Xenopoulos, K. Fadgen, J. Murphy, S.J. Skilton, H. Prentice, M. Stapels, and W. Chen, mAbs4, 24–44 (2012).
- K. Bomans, A. Lang, V. Roedl, L. Adolf, K. Kyriosoglou, K. Diepold, G. Eberl, M. Mølhøj, U. Strauss, C. Schmalz, R. Vogel, D. Reusch, H. Wegele, M. Wiedmann, and P. Bulau, PLoS One8, e81639 (2013).
- M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Biotechnol. Bioeng.29, 951–957 (2013).
- Q. Zhang, A.M Goetze, H. Cui, J. Wylie, S. Trimble, A. Hewig, and G.C. Flynn, mAbs6, 659–670 (2014).
- J.H. Thompson, W.K. Chung, M. Zhu, L. Tie, Y. Lu, N. Aboulaich, R. Strouse, and W. Mo, Rapid Comm. Mass Spectrom.28, 855–860 (2014).
- V. Reisinger, H. Toll, R.E. Mayer, J. Visser, and F. Wolschin, Anal. Biochem. 463, 1–6 (2014).
- C.E. Doneanu and W. Chen, Methods Mol. Biol.1129, 341–350 (2014).
- C.E. Doneanu, M. Anderson, B.J. Williams, M.A. Lauber, A. Chakraborty, and W. Chen, Anal. Chem.87, 10283–10291 (2015).
- A. Farrell, S. Mittermayr, B. Morrissey, N. Mc Loughlin, N. Navas Iglesias, I.W. Marison, and J. Bones, Anal. Chem.87, 9186–9193 (2015).
- J.A. Madsen, V. Farutin, T. Carbeau, S. Wudyka, Y. Yin, S. Smith, J. Anderson, and I. Capila, mAbs7, 1128–1137 (2015).
- J. Fang, C. Doneanu, W.R. Alley Jr., Y.Q. Yua, A. Beck, and W. Chen, mAbs8, 1021–1034 (2016).
- L. Huang, N. Wang, C.E. Mitchell, T. Brownlee, S.R. Maple, and M.R. De Felippis, Anal. Chem. 89, 5436–5444 (2017).
- S. Kreimer, Y. Gao, S. Ray, M. Jin, Z. Tan, N.A. Mussa, L. Tao, Z. Li, A.R. Ivanov, and B.L. Karger, Anal. Chem.89, 5294–5302 (2017).
- Z. Zhang, T. Albanetti, T. Linkous, C.J. Larkin, R. Schoner, J.B. McGivney, and N.J. Dovichi, Electrophoresis38, 401–407 (2017).
- D.E. Walker, F. Yang, J. Carver, K. Joe, D.A. Michels, and X.C. Yu, mAbs 9, 654–663 (2017).
- S. Heidelberger and K. Jonakin, The Column13, 14–16 (2017).
- S. Heissel, J. Bunkenborg, M.P. Kristiansen, A.F. Holmbjerg, M. Grimstrup, E. Mortz, T. Kofoed, and P. Horjup, Protein Expr. Purif.147, 69–77 (2018).
- C. Doneanu, J. Fang, Y. Alelyunas, Y.Q. Yu, M. Wrona, and W. Chen, J. Vis. Exp. 17, 55325 (2018).
- F. Yang, D.E. Walker, J. Schoenfelder, J. Carver, A. Zhang, D. Li, R. Harris, J.T. Stults, X.C. Yu, and D.A. Michels, Anal. Chem.90, 13365–13372 (2018).
- B. He, N. Tait, and F. Regnier, Anal. Chem.70, 3790–3797 (1998).
- F. Regnier, J. High Resol. Chromatogr.23, 19–26 (2000).
- J.H. Knox, J. Chromatogr. A.960, 7–18 (2002).
- P. Gzil, N. Vervoort, G. Baron, and G. Desmet, Anal. Chem.75, 6244–6250 (2003).
- W. De Malsche, H. Gardeniers, and G. Desmet, Anal. Chem. 80, 5391–5400 (2008).
- W. De Malsche, J. Op de Beeck, S. De Bruyne, H. Gardeniers, and G. Desmet, Anal. Chem.84, 1214–1219 (2012).
- J.C. Silva, M.V. Gorenstein, G.Z. Li, J.P.C. Vissers, and S.J. Geromanos, Mol. Cell. Proteomics 5, 144–156 (2006).
- M.R. Wilson and S.B. Easterbrook-Smith, Biochim. Biophys. Acta1159, 319–326 (1992).
Jonathan Vandenbussche is Associate Scientist LC–MS at the Research Institute for Chromatography (RIC, Kortrijk, Belgium).
Geert Van Raemdonck is Field Support Expert at PharmaFluidics (Ghent, Belgium).
Jeff Op de Beeck is Principal Scientist at PharmaFluidics.
Jenny Ho is Proteomics Application Specialist at Thermo Fisher Scientific (Hemel Hempstead, UK).
Andrew Williamson is Field Application Specialist at Thermo Fisher Scientific (Hemel Hempstead, UK).
Aran Paulus is LC–MS Integration Program Manager at Thermo Fisher Scientific (San Jose, California, USA).
Paul Jacobs is Chief Operating Officer (COO) at and co-founder of PharmaFluidics.
Pat Sandra is President of RIC and Emeritus Professor at Ghent University (Ghent, Belgium).
Koen Sandra is the editor of “Biopharmaceutical Perspectives”. He is Scientific Director at RIC and at anaRIC biologics (Ghent, Belgium) and Visiting Professor at Ghent University. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column should go to the editor-in-chief, Alasdair Matheson, at amatheson@mmhgroup.com in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence about this column should go to the editor-in-chief, Alasdair Matheson, at amatheson@mmhgroup.com







![LCE0918_Liu Bio[2]-1.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Fbdc67a7da979cf6ba6cf989305aae36e6b3a9575-200x262.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "LCE0918_Liu Bio[2]-1.jpg")






Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.






