Minimizing Fluctuating Peptide Retention in 2D-LC: How to Address a Moving Target
LCGC Europe
Retention of peptides is strongly dependent on solvent composition in reversed-phase separations with gradient elution. In this instalment we provide tips, tricks, and suggestions for best practices to help minimize retention time variations over time.
Retention of peptides is strongly dependent on solvent composition in reversed-phase separations with gradient elution. In this instalment we provide tips, tricks, and suggestions for best practices to help minimize retention time variations over time.
At a conference I attended in the summer of 2019, I listened to Dr. Patrik Petersson share results from his work on the development of two-dimensional liquid chromatography (2D-LC) methods for the characterization of therapeutic peptides and related impurities. A significant part of his talk was focused on the need to take special precautions in mobile-phase preparation and operation of the LC system to reduce retention-time variation, particularly in the first dimension of the 2D-LC system. Some of his suggestions are routinely recommended by instrument manufacturers, and will be familiar to many users. However, some of the other suggestions will not be so familiar, so I’ve asked Patrik to summarize his deep experience in this area in a way that captures all of these tips and tricks in one place. Minimizing retention variation is particularly important in some types of 2D-LC separation, but the strategies presented will also be useful to anyone looking to obtain more consistent separations using conventional one-dimensional (1D)-LC systems as well.Dwight Stoll
Heart-cutting 2D-LC is an offâtheâshelf technique offering important advantages over conventional 1D-LC separations. For example, unknown molecules eluted from separations involving salty mobile phases can
be characterized-nearly in real time-by transferring them to a second dimension separation running with a mass spectrometry (MS)-compatible mobile phase and directly into a mass spectrometer (1,2). In other situations, combining different selectivities in the two dimensions (1D and 2D) provides a possibility to significantly increase resolution compared to what can be achieved with a single column. This selectivityâbased approach to increase resolution is already available today, and can serve as an alternative to increasing resolution by increasing chromatographic efficiency of a single separation. The latter would require smaller particles in capillary columns, and an entirely new type of LC system capable of handling extremely high pressure and dissipation of frictional heating (3). Another approach to increase efficiency is to use very long columns packed with large particles and a very low flow; however, that approach is not very practical, nor popular because it requires very long analysis times (4,5).


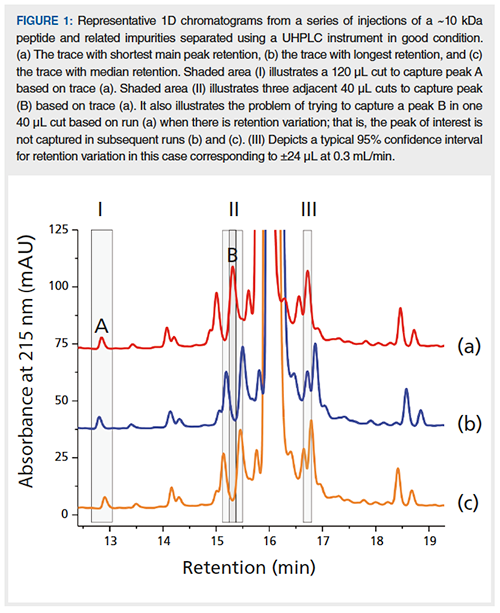
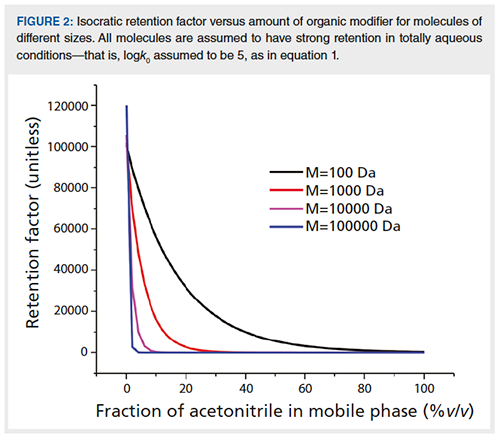
Heart-cutting 2D-LC works very well for small molecules. It also works for larger molecules, such as peptides and proteins. However, for larger molecules, it can be more challenging due to 1D retention fluctuations resulting in a moving target-that is, trapping peak(s) of interest can be a challenge, as illustrated in Figure 1(a) to 1(c) (II), where a 40 µL cut defined based on a 1D analysis (a) would miss its target in subsequent analysis (b) and (c). These fluctuations are related to a very strong response of these large molecules to small changes in mobileâphase composition. Snyder and co-workers have shown that the following expression is valid for reversed-phase chromatography of large molecules such as peptides and proteins (6):
logk = logk0 + 0.25 (M)0.5 Φ [1]
where k is the isocratic retention factor, M is the molecular weight of the analyte, and Φ the fraction of organic modifier in the mobile phase; k0 is the retention factor with no organic modifier in the mobile phase. As shown in Figure 2, this means that the retention of large molecules can change dramatically in response to even small changes in mobile-phase composition. Thus, small variations in the composition of mobile phase delivered by the pump to the column can result in practically significant variations in retention time for large molecules that would not otherwise be noticed for small molecules.

Since a modification of one functional group in a large molecule can result in a relatively small change in physical and chemical properties of the molecule compared to the same modification of a small molecule, it is also more challenging to separate related impurities for large molecules. Consequently, large molecules require very shallow solvent gradients in order to separate related impurities by reversedâphase LC. In our work, we often have LC purity methods for peptides with a slope of about 0.2%/min at a flow of about 0.3 mL/min, and still chromatograms typically show a cluster of poorly separated impurities around the main peak, such as that shown in Figure 1.
One solution to the problem could be to use peak-based capture of cuts (that is, cuts defined by a change in UV signal slope, or rise above a threshold absorbance). This works nicely for well isolated peaks, but for peptide impurity analysis this approach is less viable due to the sample complexity and poor separation. Therefore, peptides and proteins are typically analyzed using time-based cuts.
In this “LC Troubleshooting” column, I present what I believe to be current best practice to address the problem with 1D retention variation. The recommendations are based on four years of experience working with 2D-LC within the biopharmaceutical industry. Some of the recommendations are based on hard data, and some are less well founded.
Strategies to Minimize Pump Related Contributions
In our experience, it is important to use a high-pressure mixing pump (typically a binary pump) in the first dimension of a 2D-LC system to obtain the best possible retention stability. In a recent head-to-head comparison of stateâof-the-art ultrahigh-pressure liquid chromatography (UHPLC) instruments conducted in our laboratories, we found that a low pressure mixing pump (typically a quaternary pump) gave 2.4-times higher retention variation than a high pressure mixing pump from the same vendor (four systems of each type were tested on two occasions over a period of six months). Another conclusion from this work was that high pressure mixing pumps from three different vendors produced results with very similar retention variation. For a 6 kDa peptide and an LC system in good condition, the retention variation was approximately ±0.2% relative standard deviation (RSD) for a 0.15%/min reversedâphase LC gradient at 0.3 mL/min (four systems of each type tested on two occasions over a period of six months).

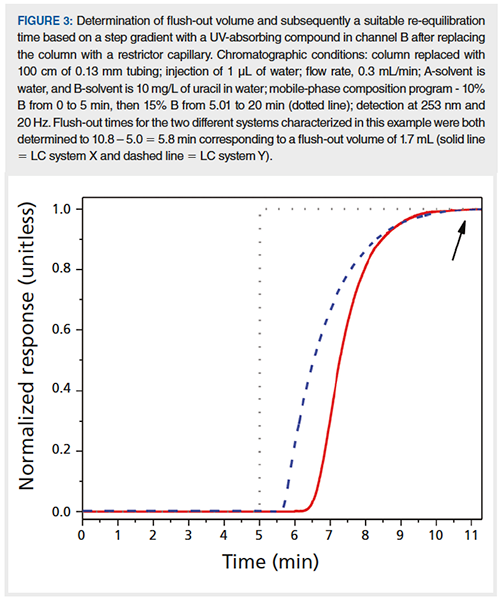
It is also important to have sufficient re-equilibration time between gradients. In the previously mentioned head-to-head comparison of UHPLC instruments, we found that the retention variation could be reduced by a factor of three by simply increasing the re-equilibration time to match the flush-out volume of the system; that is, the volume it takes for the mobile phase composition delivered to the column inlet to reach steady state after a programmed step change in composition. To determine the mixing characteristics of the 1D system and thereby define a sufficient re-equilibration time, we use the approach illustrated in Figure 3 and the following equation:
teq = (5 Vm + Vf)/F [2]
where teq is re-equilibration time, Vm is column dead volume, Vf is flush-out volume, and F is flow rate. For the high-pressure mixing UHPLC systems equipped with trifluoroacetic acid (TFA) mixers from three different manufacturers that we have characterized, we find that Vf ≤ 1.7 mL. Knowing this and that Vm is approximately 0.3 mL for a 150 × 2.1-mm reversed-phase LC column, it is possible to estimate a re-equilibration time suitable for current UHPLC systems.
If the pumping system allows definition of solvent composition and compressibility for the mobile-phase components, it is recommended to set these according to the solvents in use to obtain best possible performance.
Depletion of the volatile organic modifier from pre-mixed mobile phases results in constantly increasing retention time over a sequence of injections. To minimize this, we avoid using organic modifier in the A-solvent during 2D-LC analysis. Removing water from the B-solvent would be beneficial; however, this is often impractical, due to solubility problems associated with mixing salt-based mobile phases and organic solvents.
Twenty years ago, Dolan and coâworkers (7) addressed the problem with varying peptide retention in shallow high performance liquid chromatography (HPLC) gradients by increasing the amount of organic modifier in the A-solvent and decreasing it in the B-solvent while maintaining the slope of the gradient expressed as %acetonitrile/min. This results in an increase in %B/min, and should be less demanding because a larger volume has to be pumped to achieve a certain increase in %acetonitrile. The approach seems very logical, but when we evaluated it on four of our UHPLC systems, it did not significantly reduce the retention variation. It should be stressed, however, that we so far have only evaluated the approach for one peptide.
Another approach that we have evaluated is to scale the method for a wider column (8). The idea was that using a method with a higher flow rate would be less demanding to generate an accurate gradient.
As with the previous approach, this would require a larger volume to be pumped to maintain the same %acetonitrile/min. The approach was evaluated using both 3- and 4.6-mm internal diameter (i.d.) columns, but no improvement in retention variation was observed for the four UHPLC systems evaluated.
Other considerations that promote high pump performance include the following:
- In order to obtain the best possible homogeneity of the mobile phase delivered to the 1D column as well as reduce UV baseline noise we use large volume TFA mixers of approximately 400 µL.
- Ensure that the piston seal wash solution has been primed to wash and lubricate pistons.
- It is also helpful to monitor the pump pressure ripple and run a leak test before analyzing real samples to spot pump related problems, such as a slightly leaking piston seal, air bubbles, or a malfunctioning check valve.
Strategies to Minimize TemperatureâRelated Contributions
Variations in ambient temperature may slightly influence the system operation. This becomes prominent when analyzing large molecules, whose retention is highly sensitive to changes in temperature as well as mobileâphase composition. It is therefore important to feed the pump with solvents at a consistent temperature. The density of the solvents going into the pump changes with temperature and therefore also the composition of the mobile phase coming out of the pump. The temperature consistency of the solvent at the pump inlet is affected both by the laboratory temperature and the temperature of the liquid in the bottle. For this reason, it is an advantage to have a stable ambient temperature. The temperature control in our laboratory is tight at ±0.6 °C (maximal deviation over 24 h). To maintain this temperature stability, it is helpful to avoid direct exposure of sunlight into the laboratory. Finally, we also place the mobile-phase bottles on the system the day before usage to allow thermal equilibration after preparation (considering the length of the tubing from the flask to the pump this is probably not necessary).
Heat of friction related to compression or decompression of the mobile phase affects the temperature in both the pump heads and in the column. This change of temperature depends on flow rate and pressure. Since the pressure changes during the gradient, there will always be a fluctuation in temperature over the gradient, but eventually some kind of repeating pattern from injection to injection will be established (9). In order to achieve this, we program a sequence of gradients. Once we observe a stable retention (typically after 2–3 gradients), we define where cuts should be taken and replace the method in subsequent sequence lines without interrupting the sequence. The time must be the same for all gradients in the sequence. In 2D-LC separations, the reâequilibration time for the 1D separation often needs to be extended to account for the fact that the total analysis time for different separations will vary depending on the number of cuts made in each method. Thus, the total analysis should be adjusted to be consistent across different methods, independent of how many cuts are made. This approach with conditioning of the system also addresses other factors that influence retention, such as column priming effects (that is, saturation of slowly equilibrating active sites on the column). Note that the conditioning needs to be repeated even if leaving the system pumping isocratically between sequences for a while.
To obtain a stable retention, it is of course also important to have an efficient column thermostat with preâcolumn heat exchanger configured for the flow rate to be used.
Strategies to Minimize Mobile Phase Related Contributions
Thorough mixing of the mobile phase during preparation should be applied to ensure the solution is homogeneous before connecting the bottle to the pump inlet. In practice, we continue to stir our solvents with a significant vortex for several minutes, even after any salts have been dissolved.
After changing solvents on the pump, the system is extensively primed. As a rule of thumb, we use five times the volume to be replaced in the sinker, tubing, and degasser (in total, ~30 mL depending on system). In order to refresh the system (for example, when it has been pumping at low flow overnight), we flush with five times the volume in the degasser (~10 mL) in order to compensate for changes in composition that may take place in the degasser (that is, loss of volatile additives like TFA or organic modifier if pre-mixed solvents are used).
To minimize problems related to evaporation of volatile solvent components from the mobile phase bottles, we also use caps with oneâway valves that allow air to enter the flask, but do not allow vapour to exit.
Other Ways to Reduce the Problem with Varying Retention Specific to 2D-LC
For well-separated peaks, it is possible to use peak-based cuts-that is, a cut is triggered by a certain threshold or slope in the detector signal. In this mode of operation, small fluctuations in retention are not critical. However, as mentioned above, related impurities are usually poorly separated around the main component and therefore peak-based cuts are usually not helpful for peptides. For well-separated peaks, another alternative is to use a very wide time-based cut (Figure 1, cut I A). This does, however, usually require an on-line dilution with a weak solvent to focus the analyte at the head of the 2D column (2).
For poorly separated peaks, another approach is to place two adjacent narrow cuts bracketing the peak of interest as shown in Figure 1, cut II. This increases the probability that the peak of interest (Figure 1[a], cut II B) is captured in one or other of the two cuts in a subsequent analysis (Figure 1[b] or 1[c], cut II).
Sometimes, it is also possible to reduce retention instability by increasing the slope of the gradient slightly. Determination of what constitutes an acceptable increase in slope is done by an iterative approach where the gradient range for the critical step (that is, change in %B) is increased during a few injections while monitoring resolution to ensure that selectivity and resolution do not change too much.
Summary
For large (bio)molecules, successful operation of current instrumentation for heart-cutting 2D-LC requires some precautions to minimize 1D retention variation. However, this is a reasonable price to pay for the tremendous benefits of 2D-LC for some applications, such as the ability to obtain nearly real time LC–MS identifications of unknowns, even for 1D separations involving salt-containing mobile phases. In this instalment of “LC Troubleshooting”, I have presented some tips and tricks that I believe reduce or circumvent the problem. Some might also be applicable for conventional 1DâLC, if stable retention times are important to a particular application.
References
- P. Petersson, K. Haselmann, and S. Buckenmaier, J. Chromatogr. A 1468, 95–101 (2016).
- D.R. Stoll, K. Shoykhet, P. Petersson, and S. Buckenmaier, Anal. Chem. 89(17), 9260–9267 (2017).
- J.E. MacNair, K.C. Lewis, and J.W. Jorgenson, Anal. Chem. 69(6), 983–989 (1997).
- G. Desmet, LCGC Europe 21(6), 310–320 (2008).
- U.D. Neue, LCGC N. Amer. 27(11), 974-983 (2009).
- L.R. Snyder and J.W. Dolan, High Performance Gradient Elution (John Wiley & Sons, Hoboken, New Jersey, USA, 2007), p. 230.
- D.H. Marchand, P.-L. Zhu, and J.W. Dolan, LCGC N. Amer. 14(12), 1028–1033 (1996).
- P. Petersson, M.R. Euerby, and M.A. James, LCGC N. Amer. 32(8), 558–567 (2014).
- A.P. Schellinger, D.R. Stoll, and P.W. Carr, J. Chromatogr. A 1192, 41–53 (2008), Figure A.2.
Patrik Petersson is a Principal Scientist at Novo Nordisk A/S, in Copenhagen, Denmark.
Dwight R. Stoll is the editor of “LC Troubleshooting”. Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota, USA. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 60 peerâreviewed publications and four book chaptersin separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com







![LCE0918_Liu Bio[2]-1.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Fbdc67a7da979cf6ba6cf989305aae36e6b3a9575-200x262.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "LCE0918_Liu Bio[2]-1.jpg")





A Novel Two-Step Workflow for Extracting Clean Mass Spectra in LC×LC–HRMS Data
March 3rd 2025LCGC International spoke to Paul-Albert Schneide and Oskar Munk Kronik about the development and application of a novel two-step workflow—mass filtering (MF) combined with multivariate curve resolution (MCR)—for extracting clean mass spectra from trace-level compounds in LC×LC–HRMS data.



