The Future of Drug Lead Development and Analysis
LCGC North America
This month's installment of "MS - The Practical Art" reprises a talk from Chris Lipinski that examines the analytical road ahead in drug discovery and the quest to expand the chemistry landscape and its inherent analytical demands.
As I was deciding which talks from the 2006 Conference on Small Molecule Science to summarize for this column, I noted that, purely by chance, the opening and the closing talks presented a logical symmetry. The closing talk given by Charles McEwen, a highly regarded DuPont scientist, considers readily available adaptations of mass spectrometry technology he has developed to solve a diversity of problems and is planned for a succeeding column. This month's installment of "MS — The Practical Art" reprises Chris Lipinski's talk, from the opening session chaired by Michele Kelly of Pfizer, which examines the analytical road ahead in drug discovery and the quest to expand the chemistry landscape and its inherent analytical demands.



Michael P. Balogh
Retired from Pfizer (Pfizer Global R&D, Groton, Connecticut), Lipinski is now a science advisor for Melior Discovery (Exton, Pennsylvania). Many will recognize his name, as they will have heard him speak of the highly regarded "Rule of Five" that characterizes the majority of successful drugs in the market today.
Chemistry structures for all drugs are constrained by fundamental principles. However, Lipinski develops an issue that underpins the discovery process: the tendency for medicinal chemists and biologists to disagree.
Biologists have targets they love — wonderful targets from a biological perspective like protein–protein interactions. But the hit rate in screening for many of those targets is absolutely horrible, and that frustrates the biologists. In some ways, [they seem to] take out their frustration on the chemists, characterizing them as an uncooperative and stubborn lot, too fixated on rules, principles, and what they believe it takes to make a good drug.
Now if we could expand the properties base of the compound, perhaps we would succeed against these very difficult and important targets. But instead, we have a battle between biologists and the chemists, and the chemists say, 'No, given what we currently know, we can't succeed against that target.' In some organizations, depending on cultural conditions, the biologists prevail. The properties of a real, orally active, drug-like compound, resonates with medicinal chemists.
About Figure 1, Lipinski says — perhaps surprising to some of us — that regarding drug-like properties, the leads usually are inferior in all three areas. For example, a chemist might start out with a 1 μM lead compound and try to optimize its potency. Of course, he or she would like to improve the compound's solubility and permeability as well. If the chemist focuses on potency alone, optimizing the compound from 1 μm to, say, 10 nM — it now falls somewhere below the surface on the graph. So the chemist makes a wonderful ligand — very potent but not useful if not orally active. If the business plan under which the chemist was working were to develop an orally active compound, that compound would fail to make money, and just as significantly, it also would do nothing to help patients. Unfortunately, this scenario is extremely common. Generally, when you improve in vitro potency only, you not only fail to improve permeability and solubility, but you often worsen those properties — that is, what would make it a good orally active drug candidate.


Figure 1
Drug Candidates and Potency, Permeability, and Solubility
The chemists ask how soluble compounds must be. With an average-potency compound of about 1 mg/kg going into the clinic, with average permeability, you need about 50-μg/mL thermodynamic solubility. Only then can a compound be dealt with in pharmaceutical sciences with minimal formulation issues. So if the permeability of a peptidomimetic being developed as a protease inhibitor were in the lower-tenth percentile, but still at a potency of about 1 mg/kg, some 200 μg/mL would be required for thermodynamic solubility. A chemist might decide that a very high potency and excellent permeability would justify solubility as low as 1 μg/mL. Yet it is not easy to achieve such potency and permeability conditions. Some 80% of the compounds going into the clinic can demonstrate potency of only about 1 mg/kg. Translated into micromolar range, nearly all of the solubility action occurs between the high end (about 100 μM) and the low end (about 1 μM). A compound whose solubility approaches the 100 μM range is fine. But one whose solubility falls below 1 μM might prompt a chemist to seek a formulation change.
The Rule of Five
Compounds are given International Nonproprietary Names or United States Adopted Names at the beginning of Phase II efficacy studies. Selecting just those excludes compounds that failed preclinically or in Phase I: that is, we eliminate from our example compounds that exhibit atrocious solubility, bad permeability, and overt toxicity.
Lipinski plotted the distribution for four shared properties among this group: lipophilicity, molecular weight, hydrogen bond acceptors (counting the nitrogen and oxygen atoms in the compound formulas), and hydrogen-bond donors (counting OH and NH functional groups). All these plots are asymptotic. Looking at the red curve for almost 7500 compounds (Figure 2), most of which are orally active, he notes that 90% have 10 or fewer hydrogen-bond acceptors. The dark green curve represents hydrogen-bond donors, about 90% of which have five or fewer hydrogen bonds. Roughly 90% have a molecular weight (divided by 100 to keep it on the scale) of 500 or less. Furthermore, as indicated by the Moriguchi logp, the cutoff value (below which about 90% of these oral compounds reside) is about 4.15. Using one of the more modern calculation programs like ACDlabs (Toronto, Ontario, Canada) logp puts the value at 5.

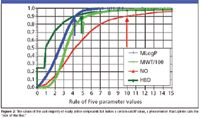
Figure 2
The values of the vast majority of orally active compounds fall below a certain cutoff value, a phenomenon that Lipinski calls the "rule of five" mnemonic. The rule of five means that poor absorption or permeation is more likely when a compound is bound by these four rules:
- more than five hydrogen-bond donors must be present;
- its molecular weight must be over 500;
- its C-log (the calculated logp value) must be greater than 5;
- the sum of nitrogen and oxygen must be greater than 10.
Note that the attributes total only four. Yet Lipinski calls his rule the "rule of five." "Well," he says by way of explaining, "the target audiences are medicinal chemists. You've got to keep things simple, easy for them to understand! Simply put everything as a factor of 5."
As in all of science, we must bear in mind some important exceptions to Lipinski's rule, which is based upon physical chemical properties; that is, the rule applies when the issues of absorption and permeation relate to the physicochemical property of a structure. If the issues relate to some kind of biological transporter, the rule does not apply. Substrates for transporters and the other natural products are exceptions. Lipinski is quick to admit that his rule does not apply in certain therapeutic areas. One is infectious disease compounds, of which many are orally active and depend upon a biological transporter. Natural products are also exceptions, which Lipinski attributes to an eons-old evolutionary selection process in which bacteria continuously struggle to prevail in their environment. All microbial warfare is chemistry-based, with each side seeking to poison its neighbors and, thus, create an environmental niche for itself. The evolutionary processes, carried out over hundreds of millions of years, have yielded exceptions, for example, orally active natural products. We do not know why these compounds are absorbed, just that evolution created them as exceptions. Lipinski goes on to point out something important about rules and common sense: "Rules play to probability, and exceptions will always exist."
Today's Reality in Oral Drugs
Properties are worse in early discovery. Approved drug properties are stable. Properties improve through each clinical stage, and the average molecular weight at approval is actually somewhat below 350; in fact, it is 347. All authoritative publications in the field agree that the properties of compounds made in medicinal chemistry have gradually become worse in terms of absorption. Yet the properties of compounds approved for marketing are unbelievably stable. The cause is a restriction on the chemistry space allowed for an orally active compound. So what kind of properties can we expect from the compounds medicinal chemists are making? In some ways, it depends upon the target. And some targets are better than others, leading to nice, easy-to-analyze compounds. For example, targets like kinases, ion channels, and aminergic G-protein-coupled receptors (GPCR) should present no trouble yielding active materials that lie within the rule of five. Proteases are "druggable" — with a lot of effort. Protein phosphatases are on the fringe, and according to Lipinski, rated "barely druggable." He says, "You can get in vitro active and selective compounds. But decent oral activity is difficult because of the kind of structures found in phosphatase inhibitors. The worst targets, from a chemistry perspective, are the protein–protein interactions."
Recent Trends in Drug Lead Development
Based upon Lipinski's experience with Pfizer compounds beginning in the 1980s, most compounds screened against therapeutic targets were below a molecular weight of 500. Then, in 1989, a jump occurred. Between 20% and 25% of the compounds screened were of high molecular weight (that is, above 500), increasing to a third of the compounds screened in the early 1990s as compared with those purchased from academic sources, which exhibited no such pattern. According to Lipinski, this trend coincided with the beginning of "full blast" high-throughput screening. The leads emerging from the screens were larger and more lipophilic.
Lipinski says that one of the big recent changes — and good news for analytical chemistry — is the advent of fragment screening, the screening of low molecular weight materials (typically MW 175–225). The screening typically looks for weak activity expressed as an enzyme inhibition constant, or IC50, in the 100 μM or millimolar range. The very weak activity demands the use of techniques such as X-ray and nuclear magnetic resonance (NMR). According to Lipinski, "It is really a surprise to me that these kinds of compounds can be optimized by medicinal chemistry. Were I looking at this 10 years ago, I would have said there is no way that a medicinal chemist is going to be able to optimize the activity of something so low in molecular weight — there's just not enough stuff there to start with."
It turns out, because of the way these compounds are screened, there is enough additional information that optimization is not just possible but also successful. There is now in the literature something called the "Rule of Three." The fragments should be of a molecular weight of less than 300, a logp less than 3, fewer than three hydrogen-bond donors, and so on. Because the screened compounds are of low molecular weight, no trend is underway to exhibit poor properties, which is the case with the large compounds. In contrast to high-throughput screening's tending toward ever larger, more lipophilic compounds that can prove more difficult to analyze, this method is not freighted with the built-in bias, which makes the analytical chemistry easier.
In another study, Lipinski illustrates data available from a variety of organizations comparing, over a period of years, trends of lipophilicity in clinical candidate compounds. Merck is flat, showing absolutely no trend toward increased lipophilicity presenting a contrast to the Pfizer data where the trend is upward. Says Lipinski, "If you look at the hydrogen bond acceptor trend, it is up in the worldwide Merck compounds, and flat in the Pfizer Groton compounds. So you have two successful organizations, which in their clinical candidates have extremely different property trends. So what is the explanation? Pfizer is on one side performing high-throughput screening while Merck, in the 1965 to 1995 timeframe, is on the other side, doing everything other than high-throughput screening. The difference in the two approaches leads to very different property profiles, because high-throughput screening inherently — unless you put in some filters — selects for larger, more lipophilic compounds." We examined how "cultural differences" led to the growth of different approaches in other aspects of pharmaceutical practice in a column from 2005, which examined metabolism studies conducted at both P&G Pharmaceutical, Inc., Mason, Ohio, and Schering Plough Research, Kenilworth, New Jersey (1).
An 80% portion of investigational new drug applications (INDs) for orally administrable drugs have a potency of 1 mg/kg, and only about 10% have a potency in the range of 0.1 mg. Dose is the key factor with respect to alternatives to oral drug delivery. With a low dose in a high potency range, delivery options are numerous. But in the 200-mg range, that option is no longer available. We might expand the kind of chemistry, making the analytical jobs more difficult. But the problem is still difficult in the discovery setting. When compared, in vitro and in vivo activity do not scale linearly, and in compounds of low in vivo dose, transporters are not saturated. Lipinski says it is easy to obtain excellent in vitro activity, but doing so is extremely difficult given increasingly active compounds when attempting to translate the activity to in vivo. "So, largely, you cannot plan for high in vivo potency. It's really a matter of luck. The problem is that we don't understand transporters. Screening across a wide range of transporters is technically very difficult, and if you cannot provide chemistry guidance, you cannot logically achieve a desired result. For chemists to produce compounds with high in vivo potency, they need screens to direct them. And right now the screens are not there. Since we need assays to solve this problem, I don't think this difficulty in getting very high in vivo activity is going to change in any way quickly in the near future."
Funky, Strange Compounds
Given Lipinski's experience, he cautions we should not expect many strange structures that deviate from the rule of five unless a miracle happens and the transporter problem is solved soon, or unless something happens to make discovery of high in vivo potency possible. Poor intestinal permeability has little chance of being solved in formulation. Lipinski says, "There is academic science, of course. But in the real world, there is not a single case of a permeability problem being solved by formulation. You can solve it in chemistry with prodrugs, but doing that is technically very difficult. There are a few things in medicinal chemistry that you can do to try to solve this problem. Peptidomimetics are arguably increasing because there are more protease targets in the human proteome than we expected."
What kinds of ligands exist for genuinely innovative targets? Roughly about three ligands per new innovative target are developed per year. In the 1990s, the pharmaceutical industry saw a decline in natural products, owing chiefly to difficulty in achieving results without resorting to added chemical complexity. Says Lipinski, "I want to underscore that needless complexity of the chemistry confers no advantage. For instance, if a screen picks up a chiral hit, the first thing we do is try to put a plane of symmetry in there, so you don't need to deal with a chiral issue."
Over the years, interest in natural products declined with the rise of automated chemistry, the demise of phenotypic screening, and the decline of infectious disease research. So, what would make natural products come into the portfolio of things to screen? According to Lipinski, it would be the reversal of all those trends. Thus, if an organization stops performing automated chemistry, it could take up phenotypic screening or increase its infectious disease research. Then natural products would likely become of interest.
"Nucleotide targets are now fairly rare," says Lipinski. "As for protein targets, the chemistry is actually directed towards hydrophobic bonding. So what we need are ligands that exploit ionic and hydrogen bonding. Activity with nucleotide targets is going to develop slowly, because few screening libraries are directed against nucleotide targets."
Protein–Protein Interaction and Diversity-Oriented Synthesis
Lipinski says that "You look at the complex structures resulting from some syntheses as a chemist and say, 'Oh, my God, what can I do here?' Small, flat, polar — you look at the thing and humus springs to mind, like something from the soil in your garden. The complexity might be what saves you on the analytical side, because such structures will be slow to find their way into the lab for analysis. Chemists will look at the complexity and say 'forget it.' Of course, their reaction will frustrate the biologists terribly, because they (the chemists) cannot do anything with compounds that stick to every surface, bind every cation known to man — in other words, a chemist's nightmare. The screening success rate so far against protein–protein interactions is essentially zero. Protein–protein interactions are tremendously important from the biological side, and it is abundantly clear that normal combinational chemistry does not work here."
So if combinational chemistry does not work, perhaps some other kind of chemistry would? Lipinski says, "That can be natural product or diversity oriented synthesis, or fragment screening on allosteric sites. Diversity oriented synthesis (DOS) is making compounds that look topologically complex, like natural products except that they are made outside the evolutionary selection process. Also, making them is very satisfying chemistry. You have multiple chiral centers, well-characterized and under control. And I would say this is different from the kind of stuff that you find in the regular screening library, and the analytical challenges will be different as well."
Lipinski offers a cautiously optimistic prediction: "If diversity oriented synthesis takes off, it will have a big impact, so it is a big experiment. DOS is being tried on protein–protein interactions, and nobody knows whether it will work. But if it does, then it is going to change what the analytical chemist receives for their projects. No matter how any company tries to hold its experience with DOS confidential, if successful it will eventually leak out. Protein–protein interaction is such an important target class that everyone is going to be involved with diversity oriented synthesis. And all of a sudden, compounds will emerge that look like natural products except that they will have been made synthetically."
But what happens if DOS fails? Lipinski says, "Then business will work as usual in analytical chemistry. So this is the big unknown. I would watch this, and see what happens. If it appears that all of a sudden people are talking about diversity oriented synthesis, figure that a couple of years down the road, you are going to start getting hit with compounds that look like complex, chiral, natural products."
Dr. Chris Lipinski, a medicinal chemist with more than 32 years of experience at Pfizer, Inc., retired in 2002 holding the position of Senior Research Fellow and now serves as a Drug-Like Properties consultant to various organizations.
Michael P. Balogh "MS — The Practical Art" Editor Michael P. Balogh is principal scientist, LC–MS technology development, at Waters Corp. (Milford, Massachusetts); an adjunct professor and visiting scientist at Roger Williams University (Bristol, Rhode Island); and a member of LCGC's editorial advisory board.
References
(1) M.P. Balogh, LCGC 23(2), 136-141 (2005).






















