Flow Rate Adjustment and System Suitability
LCGC North America
This month's "LC Troubleshooting" installment examines the possible reasons your retention times could have changed. If the root cause can be identified and corrected, no adjustment in flow rate will be necessary.
I recently received a question from a reader regarding the impact of a flow rate change on a validated method. The method was for the analysis of a multivitamin product using gradient elution with a reversed-phase liquid chromatography (LC) separation. The method ran over a 35-min period at 0.7 mL/min. The reader had observed small column-to-column changes over the several years that the method had been in use, but the main concern was that the retention times had all increased recently. It was found that an increase in the flow rate to 0.8–0.85 mL/min adjusted the retention times sufficiently that the retention times specified in the system suitability parameters could be attained. The concern was how much variation in flow rate was allowed (changes in flow rate had not been investigated during method validation) and what could have caused the problem in the first place.



John W. Dolan
This month's "LC Troubleshooting" installment will examine the possible reasons the retention times could have changed. If the root cause can be identified and corrected, no adjustment in flow rate will be necessary. After reliable system operation is assured, we will look at the influence of flow rate and the allowable changes, specifically for gradient methods.
Possible Method Changes
I like to approach LC problems using a "divide-and-conquer" technique. One of the most common questions when problems occur is whether the problem is related to a change in the method itself or the LC system hardware. In each of these two categories there are several possible causes of problems. I have listed a few of these in Table I, and I am sure that other potential problem sources exist, as well.
Let's look first at possible changes in the method, then at changes in the hardware. As listed in Table I, the most likely causes of retention time differences caused by method changes are variations in the mobile phase composition, the column chemistry, or the column temperature. We'll look at each of these in a little more detail next.


Mobile phase composition changes are an obvious potential source of retention time changes. Make the easy checks first — I would replace the mobile phase with a fresh batch. Take special care if the A-solvent (aqueous phase) or B-solvent (organic phase) is premixed (for example, 5% acetonitrile in water for A and 5% water in acetonitrile for B). Buffers can be especially problematic if they are not prepared correctly. Some methods are very sensitive to small changes in buffer pH. This should have been discovered during method development and validation or from experience using the method in routine analysis. The buffer should be used in its buffering range (±1 pH unit from the pKa), should be of sufficient concentration (generally 10–20 mM is adequate), and the pH should be adjusted before organic solvent is added. If a fresh mobile phase does not correct the problem, move on to examine the column.
Column chemistry changes: Chemistry changes between columns are much less common with today's high-purity silica columns (also called Type-B silica) than with the lower- purity columns (Type-A) that were predominant 20 years ago. However, no matter how well a manufacturer works to minimize column-to-column variation, it seems like users always come up with a special compound that shows up minor differences between columns. For existing methods, the easiest way to check for column chemistry changes is to replace the column with a new one. You can check the influence of batch-to-batch changes by ordering columns from different batches. If changes in the column due to manufacturing differences are the source of the retention time problems, you should see step-wise changes in retention with a change in columns. Double-check the observed changes by running each column at least twice in a series of experiments. For example, column 1 then column 2, then column 1, then column 2, and so forth. You should get reproducible results from each column, and a reproducible change between columns if the column packing is the source of your problem.
A second source of column chemistry problems is a change in the column due to column aging. Again, this is less of a problem with Type-A columns than Type-B ones, but it can occur. The retention times might drift over the first few injections, which is common enough that I wouldn't worry about it — just run several "priming" injections before the use of the method. If retention drifts over tens or hundreds of samples, there can be a change in the column chemistry taking place as the method is run. This can result from the buildup of contaminants on the column. A purge with 25–50 mL of strong solvent, such as 100% acetonitrile, can be sufficient to regenerate the column. If the mobile phase is especially aggressive, irreversible column deterioration can occur. For example, at pH < 2, the bonded phase tends to be cleaved from the underlying silica, and at pH > 8, the silica itself might dissolve. If your method pH is outside these limits, you should expect shorter column lifetimes (accompanied with a change in retention) unless you are using a column specially designed for extreme pH. Finally, remember that all columns should be considered consumable items — they have a finite lifetime. In my experience, a column that has given me 1000–2000 injections has performed faithful service, and in some cases survival for more than 500 injections is adequate. If your column lasts less than 500 injections, you should look into the cause and correct it if at all possible.
Column temperature changes: Temperature changes are a common source of retention time changes if the column is not operated in a column oven. For reversed-phase isocratic separations, I use the rule of thumb that a 1 °C change in column temperature can result in a 2% change in retention time. The influence of temperature on gradient retention will be less. A change in temperature for both isocratic and gradient methods can cause a change in selectivity, or peak spacing, especially if the sample contains ionic components. It should be standard practice to use a column oven for all LC methods — a method that specifies "ambient" conditions is just asking for trouble.
Possible Hardware Changes
If you are sure the method is working properly in terms of the correct mobile phase, a good column, and proper temperature control, move on to the LC system hardware as a next possible problem source. Or you might want to make a quick scan through the hardware list first so as to eliminate obvious hardware failures before you look at chemical issues. It's up to you.
The first four hardware failures listed in Table I are encountered by most users on a regular basis unless a good preventive maintenance program is in place. In each case, the failure will result in a lower flow rate and, thus, longer retention times. If your problem is shorter retention times, skip these checks.


Table I: Variables that affect LC retention times
Leaks: Leaks usually can be detected by examining all the fittings for the presence of liquid or buffer residues. For hard-to-find leaks, a scrap of thermal-printer paper can be placed against the suspect fitting. This paper is highly susceptible to discoloration when organic solvent is present and can be used to detect leaks that might be missed by visual checks. Tighten any loose fittings slightly to correct a leak — if this doesn't help, replace the fitting with a new one.
Bubbles in the pump: Bubbles will cause the pump flow rate to drop and will be accompanied by pressure fluctuations. Thoroughly degas the mobile phase and purge the pump to remove bubbles and keep them from coming back. Mobile phase degassing is the single most effective practice to improve LC system reliability. Fortunately, most newer LC systems include an in-line degasser that automatically degasses the solvent prior to use.
Pump-seal failure: Seal failure is the result of normal wear of the pump seals over time. For most applications, the seals should last six months to one year, but under some conditions, seal lifetimes can be less. I recommend changing the pump seals every six months as part of a semiannual maintenance session for laboratories where the LC system is used on a daily basis. If the LC system is used only occasionally, annual pump seal replacement is satisfactory. This is one of those cases in which preventive maintenance is more prudent than waiting until the seal fails. Seal failure usually is accompanied with the shedding of seal particles, which can work their way downstream and block frits and ruin columns. Severe seal failure can result in mobile phase dripping from the drain hole just behind the inlet check valve. If you don't change pump seals on a regular basis, you should treat them like a friend of mine does her guitar strings — if you can't remember when you last changed them, now is the time.
Check valves: Check valves might or might not be a cause of regular problems, depending upon the method conditions and instrument design. A leaky or sticky check valve can result in lower than expected flow rates. With conventional ball-type check valves, acetonitrile mobile phases seem to cause more problems than non-acetonitrile solutions. Most check valves can be sonicated safely in methanol for a few minutes to remove any contaminants (be careful — some check valves fall apart when inverted). Or you can replace a problematic check valve.
These four possible causes:, leaks, bubbles, pump seals, and check valves are the most likely sources of hardware-related problems that result in low flow rates and, thus, longer retention times. The remaining two hardware problems in Table I are much less common.
On-line proportioning errors: Proportioning errors can occur for both high and low-pressure mixing systems. Errors in proportioning can be checked using a simple gradient step test (for example, see reference 1) in which water is used as the A-solvent and water–acetone as the B-solvent. This test also highlights other problems with the system, so I recommend running it during your semiannual or annual preventive maintenance session.
Flow-controller failure: Flow controller failure can occur, although I've never seen a case of this with the instruments that I use. A simple check of flow-rate accuracy can be made by setting the flow rate to 1 mL/min and making a timed collection in a 10-mL volumetric flask. The flow rate should be within ±1% (6 s in 10 min) if the system is working correctly. If the flow-rate check fails, check the first four hardware problems, and only if you find no problem with these should you consider the flow controller as a suspect. Once you've eliminated all other problems related to flow rate, you probably will need to call the manufacturer for help on correcting a flow-rate controller error.
Adjusting Flow Rate to Meet System Suitability
The United States Pharmacopoeia (2) lists changes in chromatographic parameters that can be made to adjust an LC method so that it meets system suitability. Included in this list is an allowance for a change in flow rate of ±50%. There is little risk of introducing new problems from such changes with isocratic separations. A change in flow rate for isocratic separation will change retention time, pressure, and to a minor extent, the column plate number. However, the retention factor k and, thus, selectivity α will not change:

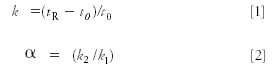
where tR is the retention time, t0 is the column dead time, and k1 and k2 are the retention factors for two adjacent peaks, 1 and 2. If flow is changed, both tR and t0 change proportionally, so k does not change and therefore α does not change, so relative peak spacing remains constant.
However, with gradient elution, the gradient retention factor k* is calculated as:

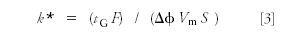
where tG is the gradient program time, F is the flow rate, Δφ is the gradient range, Vm is the column volume, and S is a constant. Selectivity for gradients is calculated in the same manner as isocratic separations shown in equation 2. Thus, you can see that a change in flow rate will change k* in gradient elution and, thus, has a potential to change selectivity. Because of this problem, the USP is revising their recommendations to indicate that changing flow rate is not recommended for gradient methods (3).
In spite of the USP recommendations, a change in flow rate from 0.7 to 0.8 mL/min, as suggested by the reader, might not change the relative peak spacing sufficiently to cause problems. This is one of those adjustments that can be justified if it allows system suitability criteria to be met. Pay special attention to the resolution between closely eluted peaks. If this peak spacing is still acceptable, a 0.1-mL/min change in flow rate should cause no other problems.
A better approach when flow rate is adjusted for gradients is to make a corresponding adjustment of one of the other parameters of equation 3 so that k*and, thus, selectivity, remains constant. For example, keep (tG F) constant. Thus, a change in flow rate from 0.7 to 0.8 mL/min would require a corresponding change in the gradient time from 35 to 30.6 min (35 × 0.7 = 30.6 × 0.8). This would guarantee no change in α, but the change in gradient time also will change the retention time, so the new retention times might still not meet system suitability. From a simplicity standpoint, it might be easier just to change the flow rate and check to be sure the separation still is adequate and system suitability requirements are met.
Summary
Let's review what we've covered. First, it is best to try to figure out why the retention times shifted. This is due most likely to a change in the method conditions or a change in the LC hardware. If a root cause can be identified, it should be corrected if possible. If the problem source cannot be found or if found, cannot be corrected, an adjustment in flow rate is justified. With isocratic methods, the proposed change is well within the USP guidelines. With gradient methods, as is the present case, a change in flow rate has a the potential to change peak spacing, so special care should be taken to ensure that peak spacing does not change. And don't forget the reminder from my quality assurance friends: "If it isn't documented, it didn't happen." Make a good record of any changes you have made to the system and include a written justification for adjustment of the flow rate.
JohnW. Dolan
"LC Troubleshooting" Editor John W. Dolan is Vice-President of LC Resources, Walnut Creek, California; and a member of LCGC's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail John.Dolan@LCResources.com
For an ongoing discussion of LC trouble-shooting with John Dolan and other chromatographers, visit the Chromatography Forum discussion group at http://www.chromforum.com.
References
(1) J.J. Gilroy and J.W. Dolan, LCGC 24(7), 662-668 (2006).
(2) United States Pharmacopoeia, 30, section 621 (2007).
(3) Pharmacopeial Forum, 34(1) (Jan-Feb 2008), in press.












New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



