Approaches to High-Speed Separations in HPLC
LCGC North America


In this installment of "Column Watch," Ron Majors examines the various approaches to increasing the speed of high performance liquid chromatography (HPLC) separations.
A current "hot topic" in high performance liquid chromatography (HPLC) column technology (1) is the alternative approaches for developing faster separations and generating more column efficiency at lower pressure drop. The big question: Which approach will be favored in the long run? These approaches include
- packed columns with small porous particles (sub-2 μm);
- packed columns with small porous particles (2–3 μm);
- monoliths (silica and polymeric);
- superficially porous packings (~2.7 μm).



Ronald E. Majors
The purpose of this installment of "Column Watch" will be to compare and contrast these various approaches for readers to assess which one might be the best solution to his or her particular separations problem.


In general, all these approaches work in balancing column efficiency, phase ratios and column permeability. The first two approaches are variations of a theme of the increased efficiency that results when the average particle size of a packed bed is reduced. As for the third approach, monoliths have been around for a decade now but have not yet achieved dominance in the applications arena. The oldest approach, superficially porous packings, dates back to the beginnings of HPLC in the late 1960s when chromatographers left the open gravity-fed columns to achieve faster more efficient separations. But the most recent versions have a new twist.


Table I: Commercial 2-mm and sub-2-mm totally porous HPLC columns at Pittcon 2007*
Small Porous Particles
Because the particles smaller than today's more common 3.0–3.5 μm sizes have the same properties, the first two categories will be combined in this discussion. Most recently, the introduction of numerous commercial HPLC columns with particle sizes in the range of 2 μm and under (Table I) has brought about a new area of controversy. As one can see in Table I, 1.5–2.0 μm particles are now available on the market. This packed-column approach uses conventional spherical silica particles but with sub-2-μm particles sizes being favored, each having various particle size distributions (2). Short columns, usually less than 50 mm in length, are run at high linear velocities giving high sample throughput. Flatter van Deemter curves for these particle sizes compared with larger sizes allow these higher flow rates without major losses in efficiency. On the other hand, if more theoretical plates are required, these small particles are packed into longer columns (up to 15 cm) but when these columns are run at these higher flow velocities, more back pressure is generated. Much has been written on this topic so I will not elaborate further. A new nomenclature has come about with terms such as ultrahigh-pressure liquid chromatography (UHPLC) has arisen to describe the higher back pressure requirement. An illustrative chromatogram in Figure 1 depicts the essence of the story. One can achieve faster separations by shortening the column and can maintain the resolution by decreasing the particle size concurrently. The evolution of HPLC is further shown in Figure 2 where plate count versus run time is plotted for various particle size columns at various lengths. The HPLC pressure limit shown on the graph is indicative of conventional pumps that can achieve pressures up to 6000 psi. New generation instrumentation allows for greatly increased pressure capability allowing the use of longer columns packed with sub-2-μm particles. In addition, high temperatures are now fashionable in HPLC to lower backpressure as well as solute improve mass transfer. In some cases with changes in temperature, chromatographic selectivity is affected in both positive and negative ways.
Figure 1
A number of "camps," mostly commercial entities, endorse porous particle sizes of 2–3 μm in diameter. Since the particle diameter is larger than the sub-2-μm particles, the pressure drop is lower but efficiency is better than the more popular 3–3.5 μm particles. Figure 3 provides a graph of the relative back pressures experienced by columns packed with various particle sizes. As expected, one can see that the 2.2-μm particles fall between the 1.8-μm and the 3.0-μm sizes. The arguments for using the packings in the 2–3 μm range are based on considering the entire separation cycle time where higher temperatures are used to improve efficiency and lower back pressure, improved liquid chromatograph hydraulics to lower band dispersion and gradient reequilibration time, and faster autosamplers, detectors, and data systems. The use of lower pressures also places less stress on the instrumentation (pump seals, pistons, check valves, and injector valve cores) and on column materials. Of course, higher operating temperatures also can aid separation speed and lower pressure drop even further for the 2–3 μm packed columns also.

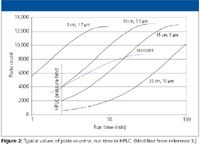
Figure 2
Monoliths
In the monolith approach, at least in the academic setting, improvements have been made in the homogeneity of the continuous beds and in efficiency. The efficiency improvement have been accomplished by adjusting the relative sizes of the macro-and mesopores, which can be varied independently using the sol-gel synthesis process, has led to better overall chromatographic columns. By varying this ratio, monolith permeability can be affected, sometimes in a detrimental way taking away one of the advantages of monoliths over packed beds — lower pressure drop. Major practical advances in silica-based monolith technology have occurred in the studies of Tanaka and coworkers from the Kyoto Institute of Technology, Japan. Recently, their work was devoted to increasing the plate count by constructing long monolithic columns, to incorporating new chemistries into the backbone for selectivity changes and to using 2D separations to increase peak capacity. Single 2.5-m monolith columns were operated at a 3.83-mm/s linear velocity and generated approximately 200,000 theoretical plates at a pressure of 45 MPa. A 4.4-m monolithic silica column was employed to separate isotopologues of benzene and toluene. By using mono-dimensional HPLC, benzene and benzene-d1 were separated. A 1.3-m monolithic silica column was to separate the 16 U.S. Environmental Protection Agency (USEPA) PAHs in a time of 72 min. Columns up to 12.4 m in length were constructed by coupling shorter columns although in the process of connecting the columns some theoretical plates were lost. The 12.4-m monolithic silica capillary column provided 1,000,000 theoretical plates for a retention factor, k, of 2.4.


Figure 3
Commercial silica monolith columns are the Chromolith (Merck KgGa, Darmstadt, Germany) and Onyx (Phenomenex, Torrance, California, based upon technology licensed from Merck). Polymeric monoliths are more prevalent since they are not covered under the restricted patent protection as are the silica-based products. Polymer monoliths are available from a number of companies (5).
Commercial silica monoliths provide rapid separations of small molecules with reasonable column efficiencies. Their efficiency falls in between the 5-and 3-μm packed columns (see Figure 2). The claim to fame of monoliths is their high column permeability (lower pressure drop) relative to the 3–5 μm packed columns. At the same linear velocity pressure drops of monoliths can be as low as 40% that of packed columns of the same efficiency. The commercial monoliths are available in internal diameters of 3, 4.6, 10, and 25 mm with a maximum length of 100 mm. The silica rods are more difficult to construct in lengths longer than 100 mm but, because of their lower pressure drops, columns can be coupled in series to gain additional plate counts. The separation of substituted benzenes on 10 Chromolith columns (1 m) plumbed in series is depicted in Figure 4. At 1 mL/min, the pressure drop across these 10 columns was only 1250 psi. Merck's silica monoliths also are available in 150 mm × 0.1 mm dimensions but these columns are prepared in situ while the silica rod columns are prepared outside of the column hardware, undergo shrinkage, and then are encapsulated in PEEK to prevent wall flow effects. To get separation speeds rivaling short porous microparticulate columns, flow rates of 3–4 mL/min are more common which is way too high for LC–mass spectrometry (MS) sources. With lower flow rate requirements, a 2-mm i.d. column would be more desirable. So far it has proven difficult to fabricate such a column dimension but work is proceeding in that direction.


Figure 4
Superficially Porous Particles
Superficially porous particles were among the first particles to appear on the scene in the early days of HPLC. Based upon the theoretical predictions of Purnell (6) and Golay (7) and experimental gas chromatography (GC) results of Horvath's Ph.D. thesis (8,9), using a support with an impenetrable, hard core coated with a thin outer layer of a porous solid should provide high-speed solute mass transfer in the stationary phase. Horvath and coworkers (10) first demonstrated the so-called porous layer bead (PLB, or pellicular) support for ion exchange chromatography. This medium became the first commercial HPLC packing that provided convincing results. Later Kirkland (11) and K. Bombaugh and J. Little of Waters developed similar products that were useful in liquid–liquid chromatography and liquid–solid (adsorption) chromatography. Compared with the large porous particles used earlier in LC, the 30–40 μm diameter superficially porous packings (1-μm phase thickness, 100-Å pores) provided much faster separations, in the 20–30 min range rather than several hours, gave more efficient separations (HETP values in the 1–2 mm range), and better limits of detection. However, because their surface areas were greatly reduced, sample capacity was limited. Since the particles were quite dense, these pellicular packings could be dry-packed using tap-vibrate methods that were used back then for packed GC columns.
The concept of superficially porous particles has been revisited several times over the four-decade history of HPLC. First, the poroshell particles initially introduced by Kirkland (12) in 1992 and commercialized in 2000 by Agilent Technologies as Poroshell were based upon the same principle as the porous layer beads but the particle size was smaller, 5 μm instead of 40 μm, and the porous layer was only 0.25 μm instead of 1 μm (see Figure 5b). The pore size of these superficially porous particles was 300 Å meaning they were more suitable for macromolecules such as proteins. The thin shell allowed the slowly diffusing proteins to only partially penetrate the packing (since the solid core prevented further diffusion). Compared with totally porous packings of the same particle size, mass transfer was more rapid and the flow rate could be run faster without sacrificing as much column efficiency (14).

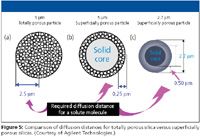
Figure 5
The most recent introduction of a superficially porous particle is the so-called fused-core particles (Figure 5c). These new superficially porous particles (~2.7 μm), a subset of the 2–3 μm particles in terms of pressure characteristics, has a solid inner core and a porous outer shell. The outer shell is sufficiently thin (0.50 μm) to allow rapid mass transfer into and out of the stationary phase; because the inner core is solid, analytes cannot penetrate any further. This diffusion path length is shorter than the porous particles of approximately the same diameter and roughly equivalent to the sub-2-μm particles. The pore size is 100 Å, which implies that the column is more suited to the separation of small molecules. Proponents here say that you get the lower pressure drop of the larger particle and the efficiency of the smaller particle. The surface area of the base superficially porous particle is 150 m2 /g. Because part of the particle consists of a solid, nonporous core, the sample capacity might be somewhat less than a typical porous particle. The particle is stated to be 50% more dense that a totally porous particle so a column-to-column comparison has the same surface area per column (15). The commercial product was developed by Advanced Materials Technology (Wilmington, Delaware) and is marketed under the tradename Halo by MacMod (Chadds Ford, Pennsylvania) and as Ascentis Express by Supelco (Bellefonte, Pennsylvania).
Due to the lower pressure drop but similar efficiency as the sub-2-μm columns, the columns can be added in series to achieve higher plate numbers. Figure 6 shows a separation of benzene and deuterated benzene in just over 12 min using three 15 cm × 4.6 mm columns plus a 10 cm × 4.6 mm column in series. Plate counts for the neutral compounds were in excess of 100,000 with a 480-bar pressure drop.


Figure 6
HPLC Separations Using Elevated Temperature
Coincidental with the interest in faster separations, the role of temperature in improving separations has come to the forefront. The use of elevated column temperatures has always been a factor to improve results in size exclusion chromatography (dissolution of polymers) and in ion-exchange chromatography (improvement of mass transfer and exchange kinetics). The use of higher temperatures has arisen mainly because of the increased pressure experienced in the newer high pressure columns that stress the upper limits of conventional HPLC systems. Mobile phase viscosity decreases with increasing temperature and, thus, column back pressure decreases. Systems with a maximum pressure capability of 400 bar can then be used with the sub-2-μm columns without over-pressuring the pump.
There are other benefits of using higher temperature. Retention decreases with increasing temperature improving separation speed, column efficiency is improved by a reduction in mobile phase viscosity resulting in faster mass transfer, and peak shapes can be improved. Since retention decreases with temperature, a lower concentration of organic modifier can be used in reversed-phase chromatography. Thus, there is less organic waste generated making high temperature separation a "greener" chromatographic technique. In addition, in some cases, temperature programming can be used as a replacement for or supplement to gradient elution. Relative retention (selectivity) can be affected by temperature changes and, thus, peaks can move in the chromatogram. Such movement can be positive (better separation) or negative (merged peaks) so the impact of temperature on selectivity is a wash.
On the downside, conventional silica-based bonded phase columns are limited in their upper temperature limits, usually to about 80 °C. However, new types of stationary phases have come onto the market that will withstand higher temperatures. Sterically protected bonded silicas, polydentate and encapsulated silica, zirconium oxide, inorganic–organic hybrids, graphitized carbon, and many polymeric materials such as polystyrene–divinylbenzene copolymers can cover the 80–200 °C range.
One of the fears of using higher temperature has been analyte stability. Indeed, compounds that are unstable at higher temperatures can degrade is left in such an environment for hours. However, at the fast flow rates used in most of the previously discussed approaches, the residence time of any particular analyte might only be a few minutes rather than hours. Studies have confirmed that for the most part, at the flow rates commonly used, analyte degradation is not a major problem. Clark (16) studied dicumyl peroxide (DCP), a dialkyl peroxide known to thermally decompose, in a reversed-phase mobile phase system of acetonitrile–water (40:60) at temperatures of 90–170 °C and flow rates of 1, 2, and 4 mL/min. She found that under many conditions, DCP did not decompose. At 130 °C, a slight degradation occurred and only at 170 °C was the degradation quite noticeable, especially at low flow rates (that might be expected). At 4 mL/min, (that is, shorter residence times), the rate of degradation was decreased with about 55% of the DCP. Some of the decomposition was attributed to the preheater tubing, which caused the DCP to degrade even before it hit the column. Further studies on several active pharmaceuticals showed no or minimal degradation at temperatures up to 100 °C.
Yan and coworkers (17) studied steroid degradation and found that decomposition occurred by interaction with the acidic surface of silica and not by temperature decomposition. With polymeric packings, they could be analyzed successfully at temperatures as high as 150 °C without any noticeable decomposition. It is interesting to note that these compounds can be analyzed by GC at this temperature without degradation so there is no reason to believe that they cannot be handled by HPLC unless other interactions (for example, acidic surface) occur.
One of the more complete studies on analyte stability in high temperature LC was conducted by Thompson and Carr (18). They investigated a number of important basic pharmaceutical compounds as well as stable organic compounds at temperatures in excess of 100 °C in aqueous mobile phase systems. Generally, they found that on the time scale of the chromatographic run, most of the compounds studied were able to withstand temperatures up to 190 °C without worry about degradation. Furthermore, they proposed criteria by which a particular analyte can be rejected as a candidate for high-temperature analysis.
Comparisons of the Different Approaches for Speeding Up HPLC Separations
All of the previously discussed approaches are directed at improving the speed of separations in HPLC. As the particle size decreases, columns can be shortened and still maintain equivalent resolution. Although the pressure drop also rises with the inverse square of the particle diameter, the pressure drop also decreases proportional to column length. So, the use of very short columns, less than 50 mm, provides good separations at reasonable pressure drops. For particles in the 2–3 μm range, column efficiency is better than the 3.5-μm columns but not as good as the sub-2-μm columns. Unless all of these short columns are run at extremely high flow rates, the excessive pressure drop is not a big consideration. For relatively simple mixtures, one can easily use both types of columns isocratically and with gradients. High temperature can be used but is not required.
For more complex samples, where a higher peak capacity is required, then longer columns are required. Commercial columns for sub-2-μm packings are available with lengths up to 150 mm, and columns with 2–3 μm particles are available up to 250 mm in length. As the column length increases, assuming all other experimental parameters stay constant, the pressure will increase proportionally. Here those approaches which can serve to decrease pressure (for example. use of monoliths, superficially porous packings, high temperature) can prove useful. Serially coupled, lower pressure drop columns can be used for increased plate counts without using excessive pressure as has been demonstrated in some earlier examples. High-pressure HPLC systems from 9000 to 20,000 psi are available to handle the longer columns packed with smaller particles.
Future Discussions on These Topics
To add to the comparisons, at the recent 31st International Symposium on Capillary Chromatography and Electrophoresis (Albuquerque, New Mexico, November 28–30, 2007), a panel discussion with the topic "Monoliths vs. Small Particles vs. High Temperature HPLC" brought together leading advocates of the various approaches to kick off audience participation reminiscent of the early days of HPLC when controversial topics were the norm and discussion sessions were lively. Space does not permit to get into the depths of the panel discussions here, but further debate will be continued at the HPLC 2008 meeting in Baltimore, Maryland, May 10–16, 2008 (www.hplc2008.org).
Ronald E. Majors
"Sample Prep Perspectives" Editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, Delaware, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgc-mag.com
References
(1) R.E. Majors, LCGC 25(9), 920–942 (2007).
(2) R.E. Majors, LCGC 23(12), 1248–1255 (2005).
(3) U. Neue, 31st International Symposium on Capillary Chromatography and Electrophoresis, Albuquerque, New Mexico, November 28–30 (2007).
(4) "Fast LC: You Want High Throughput Not High Pressure", LC World Talk, Spring 2006/International Edition (Shimadzu, Columbia, Maryland), p. 5.
(5) F. Svec, Recent Developments in LC Column Technology, Special Supplement to LCGC, June 2006, pp. 18-21.
(6) J.H. Purnell, Nature 184, 2009 (1959).
(7) M.J.E. Golay in Gas Chromatography 1960, R.P.W. Scott, Ed. (Butterworths, London, 1960), p.139.
(8) Cs. Horvath, Ph.D. Thesis, Universitat Frankfurt am Main, Germany (1963).
(9) I.Halasz and Cs. Horvath, Anal. Chem. 36, 1178 (1964).
(10) Cs. Horvath, B.A. Preiss, and S.R. Lipsky, Anal. Chem. 39, 1422 (1967).
(11) J.J. Kirkland, Anal. Chem. 41, 218–220 (1969).
(12) J.J. Kirkland, Anal. Chem. 64, 1239–1245 (1992).
(13) J.J. Kirkland, F.A. Truszkowski , C.H. Dilks Jr., and G.S. Engel, J. Chromatogr. A, 890, 3–13 (2000).
(14) R. Ricker, C. Woodward, and R.E. Majors, Amer. Lab. 36(14) 25–32 (July 2004).
(15) J. De Stefano, J.J. Kirkland, and T. Langlois, High-speed HPLC of Small Molecules and Peptides with Fused-Core Particles, Eastern Analytical Symposium, November 12–15, 2007.
(16) J. Clark, Today's Chemist at Work 13(8), 43–45 (2004).
(17) B. Yan et al., J. Sep. Sci. 30, 1672 (2007).
(18) J.D. Thompson and P.W. Carr, Anal. Chem. 74, 1017–1023 (2002).


















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

