Enhancing Phosphotyrosine Proteome Coverage Using a Combined ETD and CID Approach on a LTQ Orbitrap XL ETD
Collision-induced dissociation (CID) and electron transfer dissociation (ETD) are complementary mass spectrometric fragmentation techniques. We have used CID and ETD in different approaches to analyze tyrosine phosphorylation using a Thermo Scientific LTQ Orbitrap XL equipped with ETD.
S. Lemeer1,2, M. Zeller3, A.F.M. Altelaar1,2, T. Moehring3, S. Mohammed1,2, and A.J.R. Heck1,2,
1 Biomolecular Mass Spectrometry and Proteomics Group, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, Utrecht University,
2 Netherlands Proteomics Centre,
3 Thermo Fisher Scientific
Collision-induced dissociation (CID) and electron transfer dissociation (ETD) are complementary mass spectrometric fragmentation techniques. We have used CID and ETD in different approaches to analyze tyrosine phosphorylation using a Thermo Scientific LTQ Orbitrap XL equipped with ETD.
Reversible tyrosine phosphorylation plays important roles in numerous cellular processes and is tightly controlled by the balanced action of protein-tyrosine kinases and protein-tyrosine phosphatases. In multiple cancers, aberrant tyrosine phosphorylation has been suggested to be the underlying cause (1). Therefore the detection and site specific localization of tyrosine phosphorylation has emerged over the past years.
Low abundances and the dynamic nature of tyrosine phosphorylation together with the higher abundance of non-phosphorylated peptides cause detection of this modification to be problematic. A variety of strategies ranging from mass spectrometric (MS) based methods to phosphopeptide enrichment prior to MS have been developed (2, 3, 5). Immuno-affinity enrichment by antibodies directed against the tyrosine phosphorylated residue can be used for both tyrosine phosphorylated proteins and peptides (2-4).
Despite recent advances, the tyrosine phosphoproteome is far from comprehensive and therefore ongoing method development is still essential. Here, we utilized the LTQ Orbitrap XL ETD™ to identify the phosphorylation sites from a peptide immuno-affinity purification of pervanadate treated HeLa cells.
Materials and Methods
Nanoflow LC–MS-MS was performed by coupling an Agilent 1100 HPLC system (Agilent Technologies, Waldbronn, Germany) to a LTQ Orbitrap XL ETD mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) as described previously (6). The LTQ Orbitrap XL ETD performed a full MS scan (RP 60,000 FWHM) followed by data-dependent CID and ETD MS-MS scans with detection of the fragment ions in the linear ion trap. The decision tree method was set up to fragment all doubly charged ions, all triply charged ions above m/z 650, all quadruply charged ions above m/z 900, and all higher charged ions above m/z 950 with CID and all other precursor ions with ETD.
Results
The analytical strategy is based on the use of the two complimentary dissociation techniques collision induced dissociation (CID) and electron transfer dissociation (ETD) for the identification of the peptide as well as for the determination of the tyrosine phosphorylation site. Two approaches are compared: Fragmentation of every peptide by both CID and ETD, and the use of the most efficient dissociation technique depending upon the peptide's property such as mass-to-charge ratio m/z and charge state z (so-called data-dependent decision tree, DDDT) (7).


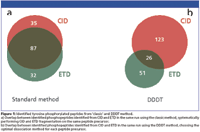
Figure 1
With the first approach 154 unique tyrosine phosphorylated peptides (Mascot score ≥ 20) were identified with the majority of sites (56%) from both CID and ETD spectra (Figure 1a). However, 35 phosphopeptides were exclusively identified with CID, whereas 32 phosphopeptides were exclusively identified with ETD. In the DDDT experiment, a total number of 200 unique tyrosine phosphorylated peptides were identified. Only 13% of the peptides (26 phosphopeptides) were identified from both CID and ETD spectra (Figure 1b). 128 phosphopeptides were identified exclusively after CID fragmentation and 51 phosphopeptides were identified exclusively after ETD fragmentation. This resulted in a 30% increase in the total number of tyrosine phosphopeptide identifications in the DDDT method, clearly indicating that this method is superior to the standard method.
Conclusion
We have shown that CID and ETD are truly complementary fragmentation techniques and their combined use greatly enhances the phosphoproteome coverage compared to MS methods that are solely based on CID. We have shown that a significant number of phosphopeptides can only be identified by ETD and the intelligent use of CID and ETD maximizes the outcome of our phosphoproteomics experiment.
References
(1) P. Blume-Jensen and T.Hunter, Oncogenic kinase signalling. Nature 411, 355–365 (2001).
(2) J. Rush, A. Moritz, K. A. Lee, A. Guo, V. L. Goss, E. J. Spek, H. Zhang, X. M. Zha, R. D. Polakiewicz, and M. J. Comb, Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 (2005).
(3) Y. Zhang, A. Wolf-Yadlin, P. L. Ross, D. J. Pappin, J. Rush, D. A. Lauffenburger, and F. M. White, Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 4, 1240–1250 (2005).
(4) P. J. Boersema, L. Y. Foong, V. M. Ding, S. Lemeer, B. van Breukelen, R. Philp, J. Boekhorst, B. Snel, J. den Hertog, A. B. Choo, and A. J. Heck, In depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immuno-affinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteomics 9, 84–99 (2010).
(5) H. Steen, B. Kuster, M. Fernandez, A. Pandey, and M. Mann, Tyrosine phosphorylation mapping of the epidermal growth factor receptor signaling pathway. J. Biol. Chem. 277, 1031–1039 (2002).
(6) M. W. Pinkse, S. Mohammed, J. W. Gouw, B. van Breukelen, H. R. Vos, and A. J. Heck, Highly Robust, Automated, and Sensitive Online TiO2-Based Phosphoproteomics Applied To Study Endogenous Phosphorylation in Drosophila melanogaster. J. Proteome Res. 7, 687–697 (2008).
(7) D. L. Swaney, G. C. McAlister, and J. J. Coon, Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959-964 (2008).


Thermo Fisher Scientific
Hanna-Kunath-Strasse 11
28199 Bremen, Germany
Website: www.thermoscientific.com















SEC-MALS of Antibody Therapeutics—A Robust Method for In-Depth Sample Characterization
June 1st 2022Monoclonal antibodies (mAbs) are effective therapeutics for cancers, auto-immune diseases, viral infections, and other diseases. Recent developments in antibody therapeutics aim to add more specific binding regions (bi- and multi-specificity) to increase their effectiveness and/or to downsize the molecule to the specific binding regions (for example, scFv or Fab fragment) to achieve better penetration of the tissue. As the molecule gets more complex, the possible high and low molecular weight (H/LMW) impurities become more complex, too. In order to accurately analyze the various species, more advanced detection than ultraviolet (UV) is required to characterize a mAb sample.



