Eluent Preparation for Hydrophilic Interaction Liquid Chromatography, Part I: Solvent Volumes and Buffer Counterions
LCGC North America


Paying attention to the details of mobile-phase preparation can have a big impact on the reproducibility of HILIC separations.
Paying attention to the details of mobile-phase preparation can have a big impact on the reproducibility of hydrophilic-interaction chromatography (HILIC) separations.
Hydrophilic-interaction chromatography (HILIC) is increasingly being used in a variety of application areas, mainly as an alternative to reversed-phase liquid chromatography (LC) when retention is too low under reversed-phase LC conditions (1). In my laboratory, we have been exploring opportunities to leverage the strengths of the HILIC mode in applications such as characterization of the highly polar metabolite fraction of plants, and more recently peptide and protein separations. In our experience many descriptions of experimental conditions reported in the literature on HILIC separations have been incomplete, or at least not explicit enough to enable analysts to accurately reproduce the results of a given study. Through conversation with a number of LC users, they report similar experiences-even those with considerable experience with HILIC separations. So, I have asked Dr. Chuck Lucy and his student Caley Craven to join my coworker Dr. Claudia Seidl and myself in preparing two installments that we hope will shed some light on the topic of eluent preparation for HILIC separations. In part I, we address the issues of solvent mixture preparation and buffer counterions. Stay tuned later in 2018 for part II in this series where we will address the issues of eluent buffer concentration and pH.
-Dwight Stoll
Introduction to HILIC Eluents
Whereas reversed-phase LC typically involves a relatively lipophilic stationary phase (for example, alkyl-modified silica) and relatively water-rich eluent (often more than 50% water), HILIC separations typically involve hydrophilic stationary phases (for example, bare or diol-modified silica) and organic solvent-rich eluent (often more than 80% acetonitrile). These variations can lead to radical differences in the retention of small molecules under the two conditions. Figure 1 shows a comparison of the separations of a simple mixture of five small molecules under reversed-phase LC and HILIC conditions. In reversed-phase LC hydrophobic molecules such as benzene and toluene are well retained and separated, but polar molecules such as uracil and cytosine are unretained (uracil is a common dead time marker in reversed-phase LC). In contrast, benzene and toluene are unretained in HILIC (toluene is a common dead time marker in HILIC), and polar molecules are well retained and separated. Although most work involving HILIC to date has focused on separations of small molecules, recently there has been considerable interest in separations of large molecules such as proteins (2). In this installment of "LC Troubleshooting," we focus on detailed aspects associated with the preparation of eluents used for HILIC separations. We encourage readers interested in other aspects of HILIC separations (for example, selectivity differences across stationary phase chemistries, and applications) to see the recent review of the field by McCalley (1).


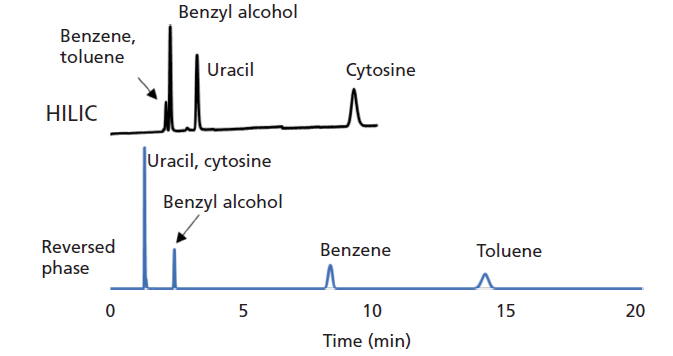
Figure 1: Comparison of retention and selectivity for a set of simple small molecule probe solutes separated by reversed-phase LC and HILIC. Reversed-phase LC conditions: 150 mm × 4.6 mm, 3.5-µm (silica) Zorbax SB-C18; mobile phase: 40:60 acetonitrile-water (premixed volumes, method A); temperature: 30 °C; flow rate: 1.0 mL/min. HILIC conditions: 150 mm × 4.6 mm, 3.5-µm (silica) Zorbax HILIC Plus; mobile phase: 90:10 acetonitrile-5 mM ammonium acetate (pH 6.8) (premixed volumes, method A); temperature: 30 °C; flow rate: 0.5 mL/min.
Although there are some examples in the literature of very well-defined procedures for preparing eluents for HILIC separations (3), it is far more common to see something like this description of the eluent: "10 mM ammonium acetate in 90:10 acetonitrile–buffer, at pH 6." When analysts go to prepare this eluent in the laboratory, they quickly confront a number of practical questions. For the sake of this example, suppose we prepare 1 L of eluent. The following questions arise:
- Is the concentration of ammonium acetate 10 millimoles in 1 L of the buffer, or 10 millimoles in 1 L of the aqueous–organic mixture?
- Was the pH adjusted or measured before or after the addition of the organic solvent to the buffer?
- Is the ratio of 90:10 on a volume (that is, v/v) or weight (that is, w/w) basis? And, if v/v, how exactly is this mixture prepared?
A list of possible approaches to answer these questions is provided later in this article.
In this installment, we tackle two aspects of eluent preparation for HILIC separations. First, we discuss some of the options for preparing the organic–aqueous solvent mixture, and demonstrate that very different results can arise from different preparation methods. Second, we demonstrate that the cation associated with an anionic buffering agent in the eluent can have a significant effect on retention as well.
Preparing the Organic–Aqueous Solvent Mixture
In previous installments of "LC Troubleshooting," John Dolan addressed this issue from the point of view of reversed-phase LC separations (4), and as part of a recent discussion of how modern LC pumps work (5). Now, we review the aspects of those discussions that are most relevant here, and extend the ideas to the impact on HILIC separations.
Taking our example of a 90:10 organic–aqueous mixture for the eluent, we can conceive of many possible ways to prepare the mixture
A. Transfer 900 mL of acetonitrile and 100 mL of aqueous buffer to a solvent bottle. These portions could be measured using graduated cylinders or gravimetrically (that is, by mass instead of volume).
B. Transfer 900 mL of acetonitrile to a 1-L volumetric flask; fill to mark using aqueous buffer.
C. Transfer 100 mL of aqueous buffer to a 1-L volumetric flask; fill to mark using acetonitrile.
Approaches A–C assume that the mixture will be prepared in a bottle, and then delivered to the high performance liquid chromatography (HPLC) column using a single channel of the pumping system. We must also consider a case involving preparation of the eluent by the pump itself.
D. Bottles of acetonitrile and the aqueous buffer are set up on the HPLC instrument, and the pump is set to deliver an eluent that is 90:10 organic–aqueous.
On the surface it appears that approaches A, B, and C would all produce the same solution; however, they will not. This difference is because of the so-called volume of mixing associated with mixing different liquids. This effect has been pointed out previously by Dolan (4,5), and others (6). In the specific case of mixing acetonitrile with water, the volume of the mixture is always less than the sum of the volumes of the constituent parts. For example, if we mix 500 mL of water with 500 mL of acetonitrile, we most definitely will not have 1000 mL of solution in our bottle-it will be less than 1000 mL (about 20 mL at room temperature). Figure 2 shows the percent loss in total volume as a function of increasing content of acetonitrile in the mixture.

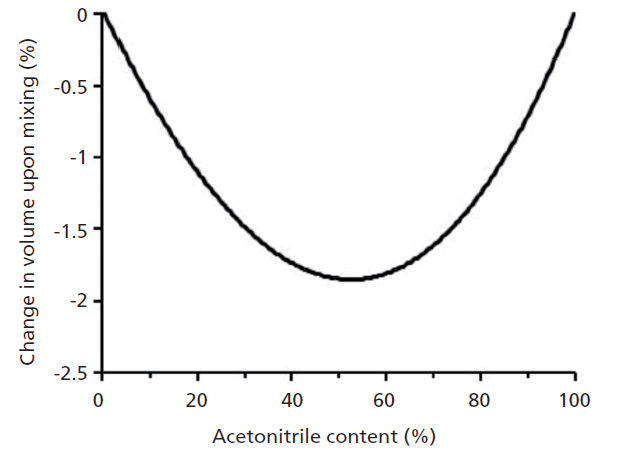
Figure 2: Percent change in total volume for mixtures of acetonitrile and water. Based on data from reference 6.
In light of this contraction in volume of mixing, let's consider our various methods for preparing our acetonitrile–water mobile phase. In method A we mix premeasured volumes of acetonitrile and water. The error in our percentage of acetonitrile in these mobile phases would be 0%. If we used method B, where we fill the flask to the mark with aqueous buffer, the final percentage of acetonitrile would be lower than we intended. Whereas making up the final volume with acetonitrile (method C) would result in a higher percentage of acetonitrile than intended. The impact of these differences can be seen in Table I, which contains experimental retention data for two neutral analytes (uracil and cytosine) and a cationic analyte (benzyltrimethylammonium [BTMA]) obtained under HILIC conditions.

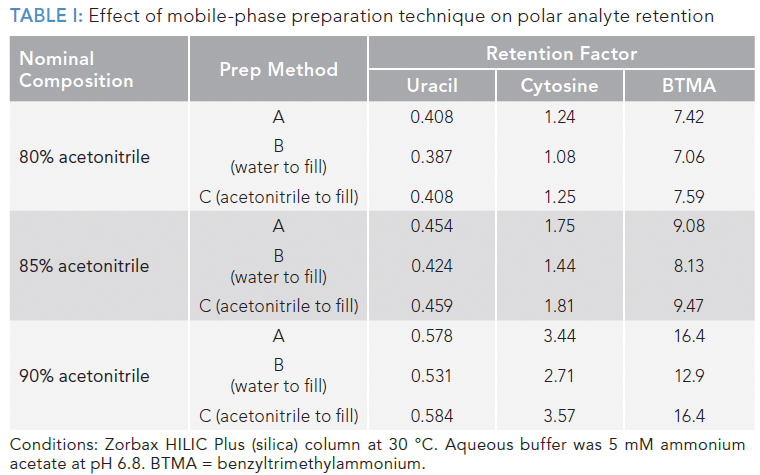
For all of the HILIC mobile phases studied here, preparing a mobile phase using method C (filling to mark with acetonitrile) resulted in slightly higher retention than expected (that is, compared to method A), whereas making the mobile phase to the mark with aqueous buffer (method B) resulted in substantially lower retention.
The moral of the story is that the exact manner in which acetonitrile and water are mixed must be described in full detail in a method for that procedure to be reproduced in another laboratory, or in the same laboratory by a different analyst.
Counterions Matter
In a future installment we will discuss how the manner in which the aqueous buffer is prepared and reported strongly affects the reproducibility of a HILIC method. Here we consider the simplest and often overlooked aspect of preparing a buffer-the counterion.
HILIC uses mobile phases with a high fraction of acetonitrile, so buffer solubility is a factor to consider. The solubility of buffers is lower in acetonitrile–water mixtures than methanol–water mixtures, and the solubility follows the trend that ammonium salts tend to be most soluble, whereas sodium salts tend to be the least soluble of commonly used salts (NH4+ > K+ > Na+) (7). Hence the popularity of buffers involving ammonium salts in HILIC work is not just because of the volatility of these buffers, making them suitable for use with mass spectrometric detectors, but also because of their solubility.
But let's consider other impacts of the buffer counterion. HILIC retention is largely due to partition of polar analytes into the water layer that forms on the surface of polar stationary phases. However, other interactions such as ion exchange and hydrogen bonding also contribute to the retention and selectivity of HILIC phases (8,9). A bare silica HILIC column will retain analytes based on both partitioning into the water layer and also ionic interactions with deprotonated silanols (–SiO-) on the silica surface. Figure 3 shows the impact of the addition of 5 mM different chloride salts to a 5 mM ammonium acetate (pH 6.8) buffer on retention for neutral (uracil, cytosine), anionic (benzenesulfonate [BS]) and cationic (BTMA) analytes, both in absolute (Figure 3a), and relative terms (Figure 3b).

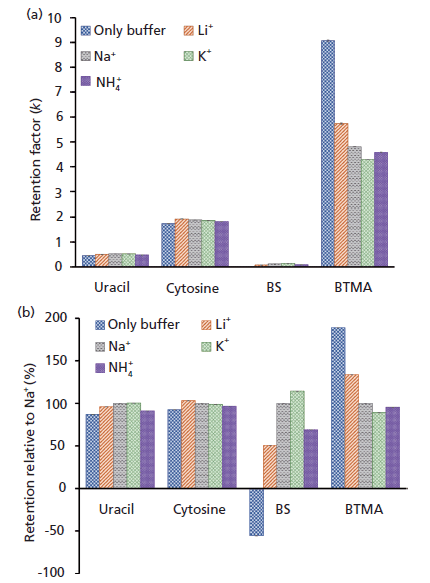
Figure 3: Retention changes due to the addition of 5 mM NH4Cl, LiCl, NaCl, or KCl to an 85% acetonitrile-15% 5 mM ammonium acetate (method A) mobile phase. Conditions: Zorbax HILIC Plus (silica) column at 30 °C.
For neutral uracil and cytosine the identity of the buffer cation causes a small but persistent shift in retention factors. For ionic analytes the impact is much greater. The anionic BS experiences electrostatic repulsion from the anionic silica surface at pH 6.8 because most surface silanols will be deprotonated and negatively charged (10). Increasing the electrolyte concentration screens the electrostatic repulsion, allowing retention of the anionic analyte to increase. The magnitude of the change in retention follows the trend Li+ < NH4+ < Na+ < K+. Cationic analytes such as BTMA undergo cation exchange with the silica surface. The addition of salt to the mobile phase provides cations that compete for the silanol exchange sites. So, the addition of salt reduces BTMA retention in a manner that mirrors the cation exchange selectivity-that is, Li+ < Na+ < NH4+ < K+.
Summary and Closing Remarks
The volume contraction that occurs when acetonitrile and water mix means that how a mobile phase is prepared affects the actual composition of the mixture in percent terms. This difference in composition, in turn, can affect the observed retention and selectivity observed under HILIC conditions. Experimental procedures for HILIC separations should describe in detail the manner in which the two solvents are mixed. One important aspect of mobile-phase preparation that is usually not described is indicating if the mixture is brought to a fixed final volume (for example, methods B and C described above), or if the measured volumes are combined to produce some nominal final volume (that is, method A above). The counterion present in the aqueous buffer also matters, particularly for HILIC separations of charge analytes.
A final bit of advice is to beware of assumptions. At the beginning of the work we did to produce the data presented here, we favored method A (where the volume of each solvent is premeasured) because it mimics how most HPLC pumps mix two separate solvents (method D). However, when we allowed our 15-year-old HPLC system to do that, we observed much higher retention than that observed with method A! Of course, this result suggests that something is not quite right with our pump (for example, one channel is not delivering solvent at the expected rate), and so we are currently putting our troubleshooting skills to work.
Acknowledgment
C. Seidl acknowledges support from the São Paulo Research Foundation - FAPESP - Process number 2016/02941-5 for her contributions to this article.
References
(1) D.V. McCalley, J. Chromatogr. A 1523, 49–71 (2017). doi:10.1016/j.chroma.2017.06.026.
(2) B. Bobály, V. D'Atri, A. Beck, D. Guillarme, and S. Fekete, J. Pharm. Biomed. Anal. 145, 24–32 (2017). doi:10.1016/j.jpba.2017.06.016.
(3) A. Socia and J.P. Foley, J. Chromatogr. A 1446, 41–49 (2016). doi:10.1016/j.chroma.2016.03.042.
(4) J.W. Dolan, LC Magazine 2(8), 582–584 (1984).
(5) J.W. Dolan, LCGC North Am. 34(6), 400–407 (2016).
(6) J.P. Foley, J.A. Crow, B.A. Thomas, and M. Zamora, J. Chromatogr. A 478, 287–309 (1989). doi:10.1016/0021-9673(89)90033-2.
(7) A.P. Schellinger and P.W. Carr, LCGC North Am. 22(6), 544–548 (2004).
(8) N.P. Dinh, T. Jonsson, and K. Irgum, J. Chromatogr. A. 1218, 5880–5891 (2011). doi:10.1016/j.chroma.2011.06.037.
(9) M.E.A. Ibrahim, Y. Liu, and C.A. Lucy, J. Chromatogr. A 1260, 126–131 (2012). doi:10.1016/j.chroma.2012.08.064.
(10) A.J. Alpert, Anal. Chem. 80, 62–76 (2008). doi:10.1021/ac070997p.
ABOUT THE AUTHORS

Dwight Stoll

Dwight Stoll is the editor of "LC Troubleshooting." Stoll is an associate professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 50 peer-reviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC's editorial advisory board. Direct correspondence to: LCGCedit@ubm.com

Charles (Chuck) Lucy

Charles (Chuck) Lucy is a professor emeritus at the University of Alberta in Edmonton, Canada. His primary research focus is the separation of charged and polar molecules using ion chromatography and hydrophilic liquid chromatography. He has coauthored more than 150 peer reviewed articles, four book chapters, and 450 presentations on analytical chemistry and separation science. He is also a contributing author to the ninth edition of Daniel Harris's popular Quantitative Chemical Analysis textbook.

Caley B. Craven

Caley B. Craven is a graduate student, at the University of Alberta in Edmonton, Canada. Currently in her masters, her primary research focus is method development for electrolytes additives in hydrophilic interaction liquid chromatography. She has just submitted her first paper and plans to continue on to her PhD after completing her masters.
Claudia Seidl

Claudia Seidl is a postdoctoral fellow in the Chemistry Department at the University of São Paulo, Brazil. She developed a study focused on column re-equilibration in HILIC under Professor Dwight R. Stoll's supervision while she was a visiting postdoctoral fellow at Gustavus Adolphus College, in St. Peter, Minnesota.










Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.

Mobile Phase Buffers in Liquid Chromatography: A Review of Essential Ideas
December 11th 2024In this installment of "LC Troubleshooting," Dwight Stoll discusses several essential principles related to when and why buffers are important, as well as practical factors, such as commonly used buffering agents, that are recommended for use with different types of detectors.


In-Loop Analyte Degradation in Two-Dimensional Liquid Chromatography: Example and Solutions
September 13th 2024In this installment of “LC Troubleshooting,” we describe an artifact that can arise because of compound degradation during the transfer of fractions of the first dimension (1D) column effluent to the 2D separation.


