Data Integrity in the GXP Chromatography Laboratory, Part IV: Calculation of the Reportable Results
LCGC North America
You need to ensure that your calculations of reportable results don’t get you into data integrity trouble. Here’s how.
This is the fourth of six articles on data integrity in a regulated chromatography laboratory. The first article discussed sampling and sample preparation (1), the second focused on preparing the instrument for analysis and acquiring data (2) and the third discussed integration of acquired chromatograms (3). In this article, we reach the end of the analytical process and focus on the calculation of the reportable results.
Scope of Chromatographic Calculations
What does calculation of the reportable results involve with chromatographic data?
- calculation of system suitability test (SST) parameters and comparison with the acceptance criteria
- calibration model and calculation of initial results from each sample injection including factors such as weight, dilution, and purity
- calculation of the reportable results from the run.
Where do these calculations take place? There are three main options to consider:
- Within the chromatography data system (CDS) application, suppliers have developed the models and tools to incorporate most calculations required by laboratories.
- Peak areas can be transferred electronically to a laboratory information management system (LIMS) or electronic laboratory notebook (ELN) to perform the calculations.
- Print the peak areas and manually enter them into a (validated?) spreadsheet or external application, such as a statistical application.
These options are shown in Figure 1. Note that we have omitted the use of a manual calculator, because this option is just too error prone for serious consideration in today's regulatory environment.


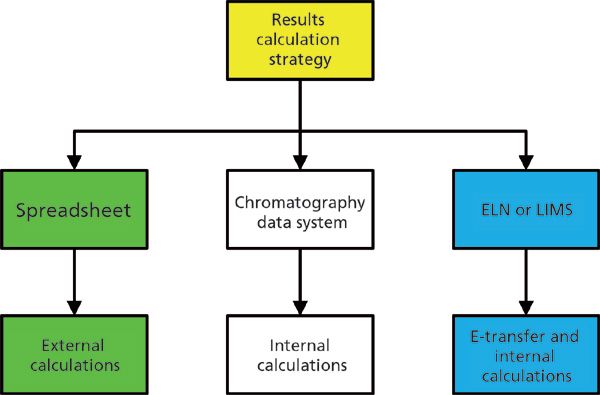
Figure 1: Options for calculation of reportable results
General Considerations for Calculations
It is worthwhile to start our discussion by defining the characteristics of calculations performed in a robust, compliant manner:
1. Calculations must accurately reflect the true state of the material under test.
2. The calculations and any changes, either before or during result calculations, must be documented and available for review.
3. Calculated values should not be seen by the analyst until the value has been recorded in permanent medium. This step includes an audit trail of the event.
4. After initial calculation, changes made to factors, dilutions, and weights used in calculation must be preserved in an audit trail.
5. All factors and values used in the calculation must be traceable from the original observations or other source data to the reportable result.
No matter where calculations happen, it must be possible to see the original data, the original calculation procedure (method), and the outcome. In addition, there must be sufficient transparency so that any changes to factors, values, or the calculation procedure are captured for review. A bonus would be an application that flags changes made to any of the above after initial use of the calculation procedure-this flag tells the reviewer that audit trails should be checked to assess the scientific merit of the change or changes.
Making a Rod for Your Own Back
Of the three options shown in Figure 1, many laboratories choose the spreadsheet. Despite the efforts of CDS application suppliers to provide a seamless way to automate calculations, many laboratories will select the least efficient and the least compliant option. Why is this? Comfort, as most people know how to generate a spreadsheet. Why bother to read the CDS, LIMS, or ELN manual and implement the option that often is the quickest, easiest, and best from a data integrity perspective? Instead, the laboratory is committed to supporting the paper industry through printing all data, entering the values manually, and employing second person reviewers to check for transcription errors.
Better Data Integrity Alternatives 1: Calculations in a CDS
The chromatography data system itself is often the best location for calculations. It usually provides access control to prevent unauthorized changes, versioning of calculations, and audit trail reviews for changes in calculated values and to the calculations themselves. In addition, the calculations are in the same system that holds the original (raw) data so that review is usually within one system. There are some data integrity risks, however:
- Some chromatography data systems will allow analysts to see calculated values (for example, plates or tailing) before they are committed to the database, permitting an analyst to process (or reprocess) data without leaving audit trail records of the activity.
- Factors, weights, and water weights (for anhydrous results) must either be entered manually or through an interface-more on this later.
- On the negative side, there are calculations that are difficult to perform directly in the chromatography system, such as statistics across multiple injections within a test run or across multiple runs. In these situations, often a spreadsheet is used to perform these calculations
Better Data Integrity Alternatives 2: Calculations in LIMS or ELN
LIMS and ELN applications, if configured correctly, generally have audit trail capabilities for most analyst actions and have the capability to perform calculations that are problematic for chromatography applications-such as the cross-run calculations mentioned above. Calculations like these are where a LIMS will shine. Access controls to prevent unauthorized actions and versioning of calculations are also available in a solid LIMS or ELN application.
On the dark side, the ability to interface is a process strength and data integrity weakness. Data sent into LIMS or ELN can be manipulated externally, then sent to the LIMS or ELN for calculation. The other weak point for a LIMS or ELN is also a weak point for chromatography calculations: manually entered factors or weights. Any time a human manages a numeric value, there is a risk that digits in that number will be transposed in about 3% of transactions (4). Another point to consider: a LIMS or ELN can perform calculations such as best-fit linear regression for standard curves, but more work is required to set up the calculations in a LIMS or ELN versus the chromatography system. In this case, the LIMS or ELN is more flexible, but the chromatography system is better adapted to a process it performs routinely.
Calculations Using External Applications
Last (and certainly least) is the humble spreadsheet. Before performing any chromatography calculations with a spreadsheet, read the "General Considerations for Calculations" section above. After reading, answer this question: Which of the general considerations is met with a spreadsheet? None, you say? You are correct. A case could be made for using a validated spreadsheet. That might be true, assuming someone doesn't unlock the sheet and manipulate the calculations (you did lock the cells and store the sheet in an access-controlled folder, correct?). Although it might be more "comfortable" to calculate results from a spreadsheet, it is worth the effort to put those calculations in the chromatography system. In addition to providing no access control or audit trail, a spreadsheet typically has manual data entry and permits an analyst to recalculate results before printing and saving the desired result values for the permanent batch record.
Some LIMS and ELN systems permit analysts to embed spreadsheets within the system, to overcome the security limitations and missing audit trail capabilities of desktop spreadsheets. If spreadsheets are to be used at all, this approach would be the preferable manner to deploy them.
A Calculation Strategy
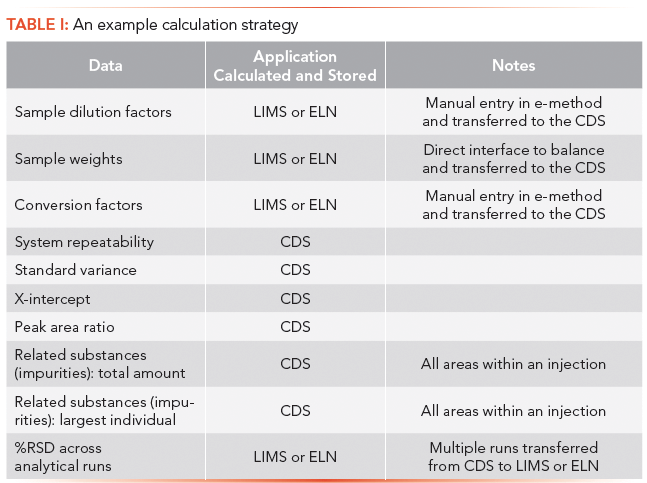
It is advisable to create a general strategy for handling calculations. The strategy should specify the types of data and where calculations are best performed. Such a strategy permits personnel at different sites to create relatively consistent electronic methods. The strategy need not be complex; it could be as simple as a table or appendix in a method development or validation procedure as shown in Table I.

Key Elements for Calculations
Wherever a calculation is used, be it from a CDS, ELN, or LIMS, the mathematical formula must be specified plus any translation into a linear string used in the application as well as the data to be used in the calculation. The data used must have the format of the data plus the ranges-for example, for pH values the format would be X.YY with an input range of something like 4.00–8.00. It is important to ensure that not just values within specification are considered but also data outside of it. The calculation needs to be tested with datasets within as well as at and outside any limits before being released for use.
Knowing how data are truncated and rounded is essential, especially when the result is used to make important decisions. The standard practice should be, "Do no truncating or rounding until the last step (which usually is the individual aliquot result or ideally the reportable result)." Before rounding, preserve all digits.
There may be scenarios where rounding or truncating of digits will happen because of technical issues, such as data reprocessing on different platforms, processors, or applications. When this adjustment happens, it is imperative that the amount of bias be understood, and its potential impact on business decisions be estimated.
Rounding and Precision of Replicates and Final Result
Reportable replicates or final results should be rounded to the same number of decimals as the specification to which they are compared. When there are multiple specifications, such as a regulatory and a release specification, then choose the specification with the largest number of decimal places for the rounding decision. In the absence of a specification, an accepted practice is to round to three significant figures.
Manipulation of Calculations
In well-controlled chromatography applications with audit trails enabled, the primary place for data manipulation is interfaces, both human and machine. Values can be manipulated outside the system, then keyed in as the correct value and used to bias reportable result values. For example, USV Ltd received a warning letter in 2014 for entering back-dated sample weights into calculations (5). Values can be manipulated outside another system-or even within that system-and then sent to chromatography using validated interfaces and accepted for processing as genuine values. In some respects, the latter situation is worse, because the manipulation is two or more steps removed from chromatography, and therefore more difficult to detect. Only by reviewing data back to the point of creation (raw data), can potential issues be detected and prevented within the analytical process.
Reporting Results
The reportable result, defined by the FDA as one full execution of an analytical procedure (6), is specified by either the applicable method or specification. Difficult questions arise when the final result meets specification but one of the replicates used to determine the reportable result is out-of-specification (OOS) or out-of-trend (OOT). These individual results are also classified as OOS and must also be investigated (6). Interestingly, most laboratories testing into compliance or falsifying results do not have any OOS results: One company had zero OOS results over a 12-year period (7). Most laboratories do not falsify results to make them OOS, unless they are very stupid.
Although OOS, OOT, and out-of-expectation (OOE) results receive attention from analysts and inspectors, it is important to recognize that falsification activities are directed at the opposite: making test results that would fail the specification into passing results through various forms of data manipulation. This probability of extra scrutiny makes it prudent to give careful review to results that are near specification limits (say, within 5%), to verify that all changes and calculations are scientifically justified.
With a hybrid CDS, most laboratories print all data rather than simply a summary, which makes it harder for the second person to review. Instead, just print a summary and leave the majority of data as electronic, thus facilitating a quicker review process (8).
Monitoring Metrics
Metrics generated automatically by a CDS application can be generated during the analytical phase of the work. The topic will be discussed in part VI of this series on culture, training, and metrics. In addition, we have written a paper that was published in 2017 in LCGC Europe on this subject of quality metrics as background reading on the subject (9).
How Can We Improve?
Direct interfaces between analytical systems can help reduce the potential for human manipulation of calculation factors. It is imperative to recognize the fundamental truth that every interface is a potential data integrity issue, and take appropriate steps to secure interfaces and the data they are transferring. One special area of concern is the system collecting the original data values: Does it permit users to create data values over and over, then select the value to be stored? Unfortunately, this is the situation for many benchtop analytical instruments and it supports the noncompliant behavior of testing into compliance. Until analytical instruments evolve in their technical design, we must rely on training, laboratory culture, and second-person observation (witness) of critical data collection to minimize the potential for this behavior (10–13).
As mentioned in the first article in this series, the preparation and recording of dilution factors is an area for improvement in integrity. Many laboratory practices still rely on analysts to manually record the dilutions that were prepared before testing. Moving these manual dilutions into procedures with gravimetric confirmation can increase confidence that dilutions were correctly prepared. It also provides a trail of objective information for investigating if result values must be investigated for failure to meet acceptance criteria or product specifications.
Is Management the Problem?
Management can create a climate where personnel are encouraged to manipulate test results. Such an environment is not created directly, but indirectly through operational metrics. Mandates like "zero deviations," "no product failures," and "meeting production targets" are each sufficient to encourage data manipulation; throw in the possibility of a demotion or dismissal for failing to meet any of these mandates and the environment is ripe for data manipulation to ensure that mandates are met-regardless of the true condition. The irony of this environment is that two losers are created: the patient who receives sub-standard product, and the company that no longer knows its true capability or process trend. This phenomenon is recognized by the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) data integrity guidance, warning that management should not institute metrics that cause changes in behavior to the detriment of data integrity (14).
Summary
Calculations and reporting are the place where raw data, factors, and dilutions all come together to create reportable values. It is critical that calculations be preserved from the first attempt to the final reported value, because of the potential for improper manipulations. This policy is essential when a second person comes to review all work carried out, as we shall discuss in the next part of this series.
References
(1) M.E. Newton and R.D. McDowall, LCGC North Am. 36(1), 46–51 (2018).
(2) M.E. Newton and R.D. McDowall, LCGC North Am. 36(4), 270–274 (2018).
(3) M.E. Newton and R.D. McDowall, LCGC North Am. 36(5), 330–335 (2018).
(4) R.D. McDowall, LCGC Europe 25(4), 194–200 (2012).
(5) US Food and Drug Administration, "Warning Letter, USV Limited" (WL: 320-14-03) (FDA, Silver Spring, Maryland, 2014).
(6) US Food and Drug Administration, Guidance for Industry Out of Specification Results (FDA, Rockville, Maryland, 2006).
(7) US Food and Drug Administration, 483 Observations, Sri Krishna Pharmaceuticals (FDA, Silver Spring, Maryland, 2014).
(8) World Health Organization (WHO), Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (WHO, Geneva, Switzerland, 2016).
(9) M.E. Newton and R.D. McDowall, LCGC Europe 30(12), 679–685 (2017).
(10) R.D. McDowall and C. Burgess, LCGC North Am. 33(8), 554–557 (2015).
(11) R.D. McDowall and C. Burgess, LCGC North Am. 33(10), 782–785 2015).
(12) R.D. McDowall and C. Burgess, LCGC North Am. 33(12), 914–917 (2015).
(13) R.D. McDowall and C. Burgess, LCGC North Am. 34(2), 144–149 (2016).
(14) PIC/S PI-041 Draft Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments (PIC/S: Geneva, Switzerland, 2016).
Mark E. Newton is the principal at Heartland QA in Lebanon, Indiana. Direct correspondence to: mark@heartlandQA.com. R.D. McDowall is the Director of RD McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com










; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.


