But My Peaks Are Not Gaussian! Part II: Physical Causes of Peak Asymmetry


Although symmetric peaks with Gaussian shapes are predicted by models of the chromatographic process, “perfect peaks” are not observed very often outside of textbooks. Several physical phenomena can lead to asymmetric peak shapes, including heterogeneity of the particle density inside the column, rearrangement of the particles over time, and accumulation of debris at the column inlet frit. Understanding these phenomena can help identify whether the cause of asymmetry is most likely to have a physical or chemical origin, which, in turn, dictates which troubleshooting steps to start with when dealing with poor peak shapes.
Last month, in the first installment of this series of LC “Troubleshooting” articles on peak asymmetry, I discussed some basic concepts in peak asymmetry, including commonly used models of chromatographic peak shapes, how to quantify peak tailing, and the impact of peak tailing on separation performance. I then went on to discuss in some detail how poorly-made connections between the column and the rest of the LC system can lead to serious peak tailing. The good news is that this particular cause of peak tailing can usually be fixed rather easily by carefully considering the parts (such as capillaries and unions, to name two) used in the flow path between the injector and detector, and replacing those that are inappropriately sized (for example, a union with a very large through-hole) or somehow improperly connected (for example, a ferrule set too shallow). In addition to these problems, there are many other potential causes of peak asymmetry—too many to cover in a single one of these articles—some having primarily physical origins, and some having primarily chemical origins. In this month’s installment, I will focus on several other potential physical causes, including problems with column packing, changes in the packed particle bed over time, and accumulation of debris in the column. A common symptom of all of the physical causes of peak asymmetry discussed here is that all peaks in a chromatogram will be affected similarly. This can be an important clue to help determine if the source of the asymmetry is more likely to have chemical or physical origins. If all of the peaks in a chromatogram are either fronting, or tailing (or both [1]), the cause is most likely physical in origin. If only some of the peaks are fronting or tail- ing, but the other peaks look good, then it is most likely that the cause of the poor peak shape is chemical in nature. This distinction is helpful when deciding which potential solutions to improve the peak shape to try first.
Brief Review of Flow Through a Packed Bed of Particles
To understand the different peak shapes that can be observed as a result of different physical problems within a LC column, it is helpful to first review some foundational concepts related to mobile phase flow through a column packed with small particles like those used in HPLC. Figure 1a shows a highly idealized illustration of the organization of particles (depicted as porous spheres) in a perfectly packed bed. Such perfect arrangements of particles are not achievable in practice, though some come close, in the case of small very small (<1 μm) particle columns (2). The main point of this illustration is that a highly ordered, consistent arrangement of the particles in the column leads to highly consistent mobile phase velocities across the column radius. Since the rate of migration of analyte molecules (that is, analyte velocity) from the inlet to the outlet of the column is proportional to the mobile phase velocity, the consistency in mobile phase velocity directly translates into consistent analyte velocities, and thus a symmetric analyte peak observed at the detector. In the case of perfectly consistent mobile phase velocity across the column diameter, there would be no “A-term broadening” of peaks, and their widths would be dictated primarily by diffusion of analytes along the long axis of the column, and into and out of pores in the particles. The consistency of the mobile phase velocity in this case is communicated in Figure 1a with yellow arrows of the same size. Analyte molecules are illustrated as green dots, and the resulting chromatographic peak observed at the detector is shown at the right of the column.

 Perfectly ordered arrangement of particles that leads to consistent mobile phase velocities and symmetric peak shapes; (b) Random arrangement of particles—local variations in the tightness of the packing lead to variations mobile phase velocity, which in turn lead to asymmetric peaks; this particular shape is referred to as a “fronting peak”; (c) Empty void space at the column inlet due to consolidation of the particle bed can lead to mobile phase mixing (orange arrows) and tailing peaks; (d) Accumulation of insoluble debris from the mobile phase at the inlet frit can lead to differences in the velocity of mobile phase passing through the frit, and thus asymmetric or even split peaks.")
FIGURE 1: Illustration of porous particles arranged in LC columns in different ways. Drawings are not to scale, are highly idealized, and with features that are exaggerated to illustrate specific concepts. Porous particles are depicted in textured circles, mobile phase velocities at different positions in the cross-section of the column are indicated with yellow arrows, analyte molecules are depicted as green dots, and the peak at the right in each case illustrates the type of peak shape that can result from the particle arrangement shown at left. (a) Perfectly ordered arrangement of particles that leads to consistent mobile phase velocities and symmetric peak shapes; (b) Random arrangement of particles—local variations in the tightness of the packing lead to variations mobile phase velocity, which in turn lead to asymmetric peaks; this particular shape is referred to as a “fronting peak”; (c) Empty void space at the column inlet due to consolidation of the particle bed can lead to mobile phase mixing (orange arrows) and tailing peaks; (d) Accumulation of insoluble debris from the mobile phase at the inlet frit can lead to differences in the velocity of mobile phase passing through the frit, and thus asymmetric or even split peaks.
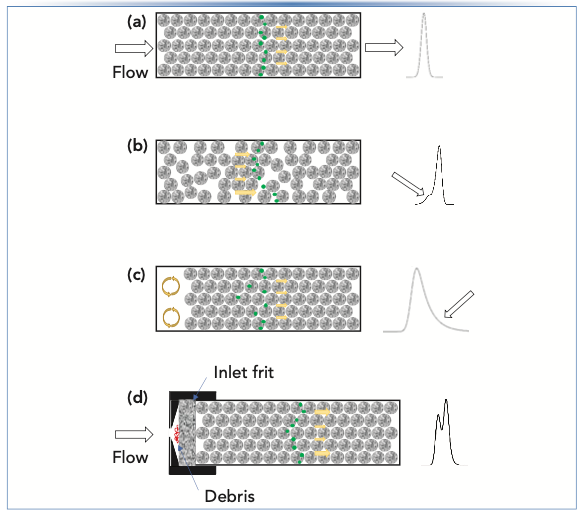
Variations in Particle Packing Tightness Throughout the Column Can Lead to Asymmetric Peaks
In contrast to the highly ordered bed structure illustrated in Figure 1a, Figure 1b shows that, in real columns, the structure of the particle bed is rather disordered, leading to regions where the particle density (that is, the number of particles per volume of column, not the density of the particles themselves) is much higher than others. In recent years, several studies discussed in the literature have made clear that much of this heterogeneity in packing density throughout the column arises as a result of friction between the column wall and the particles as they are pushed into the tube. This results in large variations in density near the wall, moving from the wall toward the center of the tube. This is sometimes referred to as a wall effect on the packing density. The net effect of this variation, though, is that the mobile phase velocity will be lower in regions of high density, and higher in regions of low density, because there is less friction opposing the flow of mobile phase as the spaces between adjacent particles increase. This variation in mobile phase velocity then manifests as an “A-term type of peak broadening,” and results in peaks that are broader compared to the situation where there is no variation in mobile phase velocity across the radius of the column. In the particular situation shown in Figure 1b, the packing density near the wall is significantly lower compared to the center of the column. If the difference in densities is large enough, this can actually result in peak asymmetry like that shown in this case, which we refer to as peak fronting. We typically do not observe this behavior much in commercial LC columns, because the manufacturers do a good job of mitigating this particular problem, but I’ve packed enough columns during my days working in the column business to know that this effect can be very serious, and must be solved through optimization of column packing procedures. During the development of new stationary phases and column technologies, column manufacturers study the impact of a large number of variables on the packing quality, including things like the slurry solvent (that is, the solvent used to suspend the particles to flow them into the column), slurry concentration (particles per volume of solvent), packing flow rate, and packing pressure. Readers interested in learning more about the column packing process and the topic of heterogeneity of the particle bed are referred to recent articles on the topic (3).
Development of Particle-Free Void Spaces in the Column Leads to Peak Tailing
When packed particle LC columns are manufactured, the slurry containing the particles is pushed into the tube at very high pressures, typically exceeding 10,000 psi for modern materials. In principle, these high pressures force the particles into an arrangement that is unlikely to change significantly when used in an LC instrument at lower pressures. In the early days of HPLC it was not uncommon for column performance to deteriorate upon settling or rearrangement of the particle bed. Fortunately, modern manufacturing procedures have generally improved the robustness of LC columns significantly, and I have to say I am often impressed at just how resilient modern columns are, surviving instrument failures (for example, pressure spiking due to sticky check valves) or user error (for example, allowing columns to dry out without flushing first with an organic/water mixture to remove buffer salts). However, rearrangement of the particle bed can still happen—for example, in response to physical stress on the column (such as repeated pressure fluctuations [4]). Although there are many ways that the particle bed could conceivably rearrange, the classical observation is that the particle bed consolidates in the direction of the outlet (think of sand settling in a bucket when vibrated), leaving a significant “void” at the column inlet where there are no particles and only mobile phase. These void spaces can then act like small mixing chambers and lead to significant peak tailing like that shown in Figure 1c. Sometimes reversing the flow direction can restore some of the column performance, but, in my experience, this performance is usually very short-lived, and when a void develops in a column it is best to just replace the column. Although many modern LC columns are highly resilient to various physical stressors, it is still helpful in the long run to avoid causes of such stress in order increase column lifetime. This includes avoiding major pressure fluctuations (for example, due to air bubbles in pumps), and properly flushing columns according to manufacturer recommendations before storing the column for more than a few days.
Accumulation of Debris on Column Frits Can Lead to Asymmetric Peaks
As I’ve written about in the past, one of my favorite troubleshooting tips is to consistently use inline filters in LC systems, especially immediately upstream from the column (5). There are many ways that insoluble debris can make its way to the column inlet, including particulate matter in the sample that is injected, particulate matter in the mobile phase that comes from unfiltered solvent feeding the pump, and polymeric material that is shed by valves (for example, from a rotor seal) like those found in autosamplers. If an inline filter is not used between the sample injector and the column, then much of this insoluble debris will accumulate on the inlet frit of the LC column. I’ve looked at many inline filters when replacing them after they’ve become blocked, and, without exception, I see that debris never accumulates across the frit in a uniform way. Sometimes it is concentrated at the edges, sometimes in the middle, and other times there is no obvious pattern. However, the non- uniform distribution of the accumulated material again means that flow through the column inlet frit will also be nonuniform, leading to different mobile phase velocity streams at the column inlet, peak dispersion, and asymmetric peak shapes. In the illustration in Figure 1d, I’ve attempted to show accumulation of debris at the center of the inlet frit, and that in some cases this can lead to severely distorted split peaks as shown at right. The good news is that this particular cause of peak asymmetry can largely be avoided through consistent use of inline mobile phase filters directly upstream from the analytical column. It is also possible in many cases to physically replace the inlet frit on the LC column, but this requires extraordinary care to avoid disrupting the particle bed, and I would not recommend this remedy as a routine practice.
Summary
In this installment of “LC Trouble- shooting,” I’ve discussed several of the common physical causes of peak asymmetry in LC, and remedies for some of them. In some cases there is not a whole lot the user can do to address the root cause of peak asymmetry, as some problems occur at the point of manufacture of the column, but in the spirit of increasing our troubleshooting knowledge, it is still helpful to know what all can go wrong, and this knowledge can also help determine if the problem is likely to be solvable, or if the column must simply be replaced. In the next installment in this series, I will discuss several chemical causes of peak asymmetry, where we as users typically have more opportunities to improve peak shape through changes in operating conditions.
References
(1) M.F. Wahab, D.W. Armstrong, and D.C. Patel, LCGC North Am. 30(12), 670–678 (2017).
(2) B.A. Rogers, Z. Wu, B. Wei, X. Zhang, X. Cao, O. Alabi, and M.J. Wirth, Anal. Chem. 87, 2520–2526 (2015). https://doi.org/10.1021/ac504683d.
(3) F. Gritti and M.F. Wahab, LCGC Europe 31(2), 90–101 (2018).
(4) E.S. Talus, K.E. Witt, and D.R. Stoll, J. Chromatogr. A 1378, 50–57. (2015). https://doi.org/10.1016/j.chroma.2014.12.019.
(5) D.R. Stoll, LCGC North Am. 35(2), 98–103 (2017).
About the Column Editor

Dwight R. Stoll is the editor of “LC Troubleshooting.” Stoll is a professor of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 75 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com


")












Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


