Analytical Methods to Determine the Stability of Biopharmaceutical Products


Analytical methods are used in the biopharmaceutical industry to ensure the quality, efficacy, and safety of drug substances and drug products. One of the fundamental measures of the quality of a drug substance or drug product, including biopharmaceuticals, is the stability of the active pharmaceutical ingredient (API). In fact, the first International Council for Harmonization (ICH) guideline on quality, ICH Q1, is for drug stability. In this column, we look at drug stability in general, differences between large- and small-molecule stability (ICH Q1 and ICH Q5), as well as the analytical methods used to measure the stability of a product. Although there are several analytical methodologies that can be used, and we touch on those briefly, we focus mostly on the chromatography and mass spectrometry methods used to characterize drug stability.
At the center of everything we do in the biopharmaceutical industry should be patients and ensuring that their medicines are high quality, safe, and effective. A cornerstone of any drug substance or drug product quality assessment is stability. Ensuring the stability of a biologic (or any pharmaceutical) gives patients confidence that their drugs are efficacious, perform as expected, and are available when needed. Stability ensures that patients receive the same high-quality product throughout the product’s expiry period (2).
Drug stability, both pharmaceutical and biopharmaceutical, can be defined as the ability of a drug substance or product to retain the same properties and characteristics, within specified limits, that it had at the time of manufacture (3). Biopharmaceuticals are complex drugs that comprise thousands of molecules and a complex amino acid chain that is folded into complex structures. As a first-year biochemistry student can tell you, that complex structure directly impacts protein function. Thus, in biopharmaceutical stability, we are looking for the correct structure of a given protein therapeutic, such that its function, or its specific therapeutic effect, is achieved. Ensuring the stability of the drug substance or product ensures a high-quality product, accurate dosing, identification of degradants, mitigation of impurity-induced adverse events, and increased patient compliance.
In the case of a biopharmaceutical, instability is most commonly referring to degradants in terms of fragments or aggregation (protein aggregation was discussed in a previous set of columns in 2015 (4,5). Product instability can lead to a loss of potency or efficacy. Degradation products can lead to impurities and contaminants and safety and toxicity issues. They can also lead to problems with immunogenicity, an unwanted immune response against oneself (6), and alterations of bioavailability. In addition, product instability can affect appearance, smell, feel, taste, and precipitation, which may impact both product efficacy and safety but also patient compliance.
This all highlights the importance of understanding drug stability. How do we characterize drug stability? To determine the stability of a biopharmaceutical, a stability testing program is developed and implemented throughout the life cycle of the product. This stability program can comprise various components, which we will not go into in any great detail. However, briefly, one might consider long-term stability testing, accelerated stability testing, annual (follow-up) stability testing, and discrepancy testing as key components of a stability testing program. Long-term testing of the drug substance or drug product serves as the basis of the recommended storage conditions and shelf-life. Accelerated stability testing provides information on degradation pathways that the product is most vulnerable to. Annual testing provides additional information to guide long-term storage conditions. Discrepancy testing can confirm the stability of specific batches. In addition to these tests, other types of studies commonly seen in stability testing are cumulative expiry (for biologics, supports processing time, temperature, freeze, thaw, and refiltration), temperature cycling, photostability, reconstitution, comparability, and in-use studies (2,7). Thus, one of the major end goals of a stability testing program is determining shelf life or expiry under specific conditions, which are determined to support the maximum stability of the drug over time. The United States Food and Drug Administration (U.S. FDA) defines an expiration date as “the time period during which the product is known to remain stable, which means it retains its strength, quality, and purity when it is stored according to its labeled storage conditions” (8).
Stability Guidelines
With a general understanding of drug stability in hand now, we can briefly discuss international guidelines for drug stability and how the differences between small (pharmaceuticals) and large (biopharmaceuticals) molecules are characterized. Over the years, there have been several guidelines developed by international consortia to individual drug authorities, World Health Organization (WHO) and others (2). Here, we focus on guidelines developed by one of those international consortia, the International Council for Harmonization (ICH), specifically the ICH Q1 series (Table I).


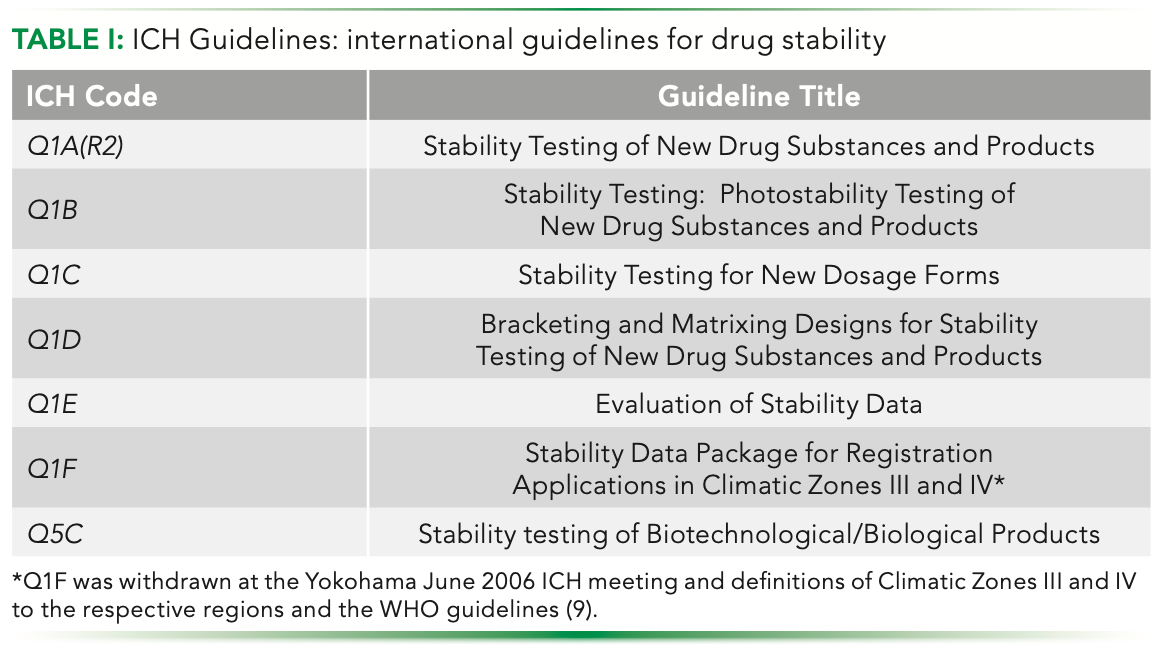
The ICH Q1 series of guidelines, like all ICH guidelines, promotes a science-based, risk-based evaluation of drug substances and products related to stability. In addition, they allow for flexibility in their implementation so that they can be broadly implemented across the world. ICH-Q1A(R2) (with the R2 indicating revision 2 of the guideline) is the foundational ICH guideline related to drug stability. The purpose of ICH Q1A(R2) is to provide basic guidance on how to ensure the quality of a drug substance or product under four general key considerations: selection of batches; study design; evaluation and outcome; and stability commitment. For the selection of batches, ICH Q1A(R2) provides guidance on what is considered a primary batch and production batch, how to select batches for specific testing, and how to consider storage conditions such as container closure. It provides guidance on formal stability studies, supporting data to be considered, how to conduct stress studies, and storage condition studies (for example, temperature and humidity), including long-term, intermediate, and accelerated testing and frequency of testing. ICH Q1A(R2) provides guidance on how to evaluate stability data and its outcomes, including specifications and significant changes. ICH Q1A(R2) also provides guidance on the appropriate stability commitment to propose and implement. It is important to note, that ICH Q1A(R2) specifically addresses pharmaceutical products (small molecule drugs), but more on that later (7).
The remaining ICH Q1 guidelines were developed and implemented in support of ICH Q1A(R2). For example, ICH Q1B was developed to address the possibility that light exposure can affect product quality. Specifically, ICH Q1B provides guidance to characterize the intrinsic photostability of new drug substances and products. It also offers guidance on the evaluation of photostability data to ensure light exposure does not result in changes to the drug substance or product. ICH Q1C, one of the shortest ICH guidelines, reiterates the application of ICH Q1A(R2) for new dosage forms. ICH Q1D provides guidance for using bracketing and matrixing in stability studies. Bracketing is defined as evaluating samples on the extremes of certain design factors at all time points. Matrixing is defined as evaluating a subset of the total number of possible samples for all factor combinations tested at a specific time point. ICH Q1E provides guidance on how to perform stability data evaluation and on such topics as extrapolation (using a known data set to infer information about future data sets) (10–13). And finally, ICH Q1F was withdrawn at the Yokohama June 2006 ICH meeting and definitions of Climatic Zones III and IV were left to the respective regions and the WHO guidelines (9). For reference, Table II provides the climatic zones used in temperature and humidity stability studies, including Zones III and IV from WHO.

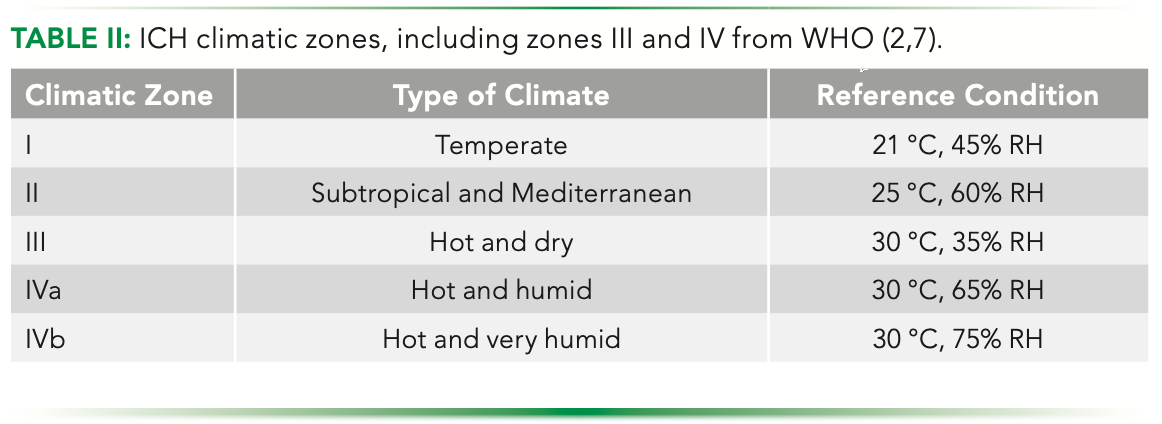
Like ICH Q1A(R2), ICH Q1B through ICH Q1E were written with pharmaceutical (small molecule) products in mind and are largely applicable to biopharmaceuticals. However, as we know, there are significant differences between large and small-molecule drugs. Briefly, more traditional pharmaceuticals are comprised of a defined chemical structure of identical copies and are synthesized chemically. There is rarely biological contamination, there are defined standards and specifications for impurities, there are sensitive, well-defined standards and discriminating analytical characterization methods, and stability programs that are well-defined and can be modeled. On the other hand, biopharmaceuticals are comprised of complex chemical structures that are comprised of thousands of atoms making up heterogeneous macromolecules made in living cells. Biopharmaceuticals are prone to biological contamination because of contamination for the production process using living cells. There are also process and product-related impurities with no fixed acceptable threshold and multiple orthogonal analytical methods used for characterization (see next section). And finally, biopharmaceuticals are susceptible to stability loss through temperature, shear, and light (14). These differences lead to the development of specific guidance, ICH Q5C, related to biopharmaceutical stability.
ICH Q5C provides guidance on stability testing of biotechnological and biological products and applies to well-characterized proteins and polypeptides isolated from tissues, body fluids, cell cultures, or if they are produced using recombinant deoxyribonucleic acid (DNA) technology. A well-characterized biologic is defined by its product quality attributes, such as appearance, purity, activity and quantity, and structure. Many aspects of stability testing contribute to elements of a well-characterized biologic, such as specifications, analytical characterization, product trending, process controls, and monitoring and adherence to current Good Manufacturing Practices (cGMP). Specifically, ICH Q5C provides specific guidance for biologics around a selection of batches, stability-indicating profile, storage conditions, test frequency, specification, and labeling. In addition, ICH Q5C in sections 5 and 8 provides guidance on test methods to detect stability-indicating profiles (15).
Analytical Characterization of Stability
To determine the stability of a biopharmaceutical, several orthogonal analytical techniques are employed for both the drug substance and the drug product. Generally speaking, these stability monitoring techniques must be able to detect changes in identity, purity, and potency. Methods are validated at the time of market approval.
For potency, ICH Q5C (15) recommends cell-based bioassays or using enzyme-linked immunosorbent assay (ELISA)-based binding assays for both drug substances and drug products. To monitor purity, ICH Q5C recommends monitoring size variants and charge variants. To monitor charge variants for both drug substance and drug product several methodologies are suggested including size exclusion chromatography-high performance liquid chromatography (SEC-HPLC), sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and capillary electrophoresis (CE)-SDS. Of note, SEC-HPLC is particularly useful in monitoring stability because it can differentiate aggregates and high molecular weight species, which are indignant of protein instability (16). Ion exchange (IEX)-HPLC and isoelectric focusing are recommended for monitoring charge state variants for drug substances and products. ICH Q5C also recommends monitoring the appearance and protein concentration for drug substance stability. For drug product stability, ICH Q5C recommends monitoring the appearance, color, clarity, protein concentration, subvisible particles (aggregates), container closure integrity, stabilizers, and preservatives.
In addition to these analytical methods, liquid chromatography–mass spectrometry (LC–MS) has become increasingly used in monitoring biopharmaceuticals and biopharmaceutical stability. A recent review by Vallejo and others (1) highlights the use of LC–MS as a tool to monitor biomolecular stability. This review gives a thorough look at MS-based techniques used to monitor protein stability from native MS to ion mobility–MS (IM–MS). However, for this column, let us review some of the trends in biotherapeutics.
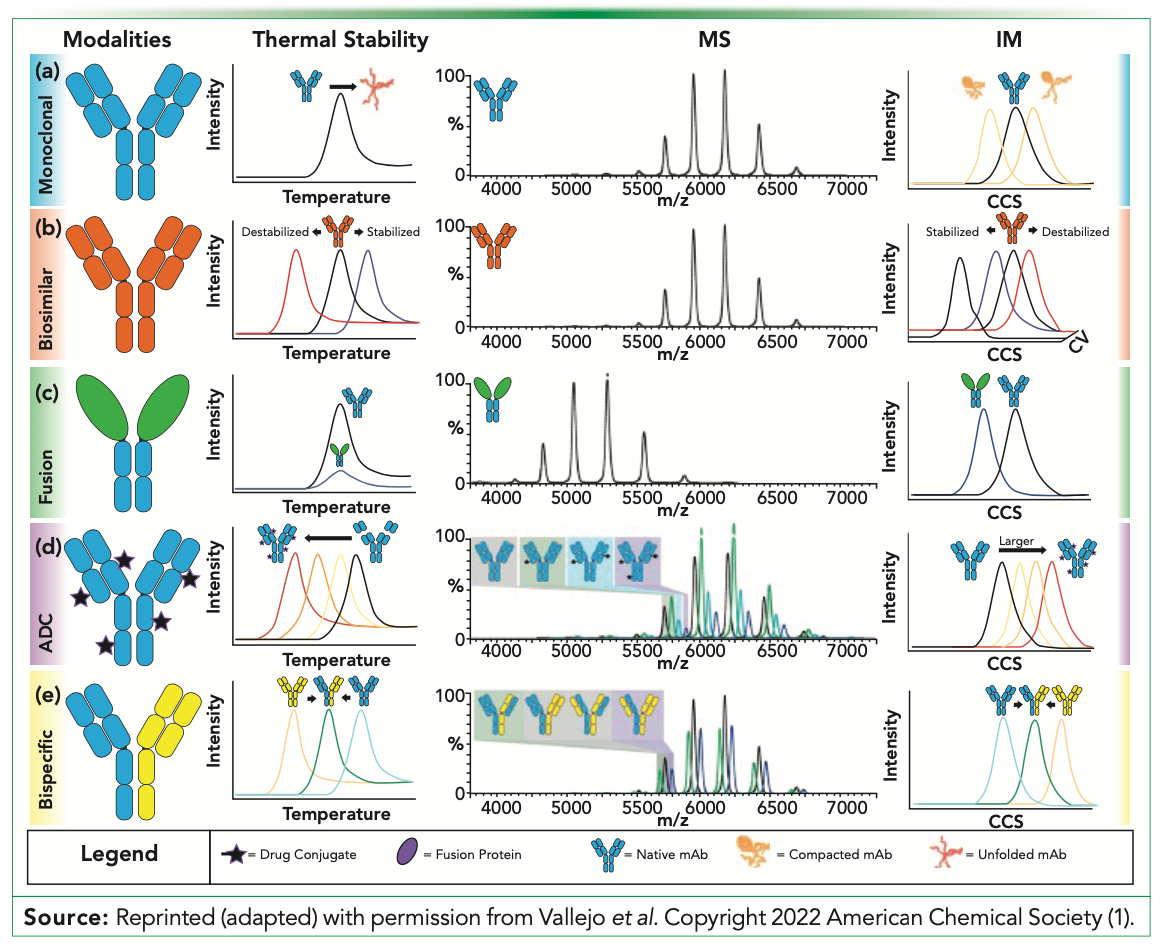
For biopharmaceuticals, in particular monoclonal antibodies (mAbs), structurally sensitive MS methods have become more widely used, critical perhaps, to rapidly analyze high molecular weight species. Some of these trends include variable-temperature MS, footprinting MS, IM–MS, and collision-induced unfolding (CIU). Figure 1 provides representative thermal stability, MS, and IM–MS stability data for mAbs, biosimilars, fusion proteins, antibody–drug conjugates (ADCs), and bispecific antibodies. Variable-temperature experiments can monitor protein stability through monitoring melting temperatures whereas gas-phase technologies, such as MS, can be used to detect changes in gas-phase mAb structures in correlation with variable temperature experiments or changes in CIU mode. In variable temperatures, MS mAb samples are heated within the ESI source and the detection of degradation products and non-native disulfide bonds can be used to characterize the mAb stability (1,17). Footprinting MS techniques, such as hydrogen deuterium exchange (HDX)-MS or covalent labeling (CL)-MS, can provide information on structural integrity, changes in high molecular weight species, and antigen interactions.

FIGURE 1: Data and information content that can be expected from variable-temperature, MS, and IM data sets for the following biotherapeutic modalities: (a) mAbs, (b) biosimilars, (c) fusion proteins, (d) antibody−drug conjugates, and (e) bispecific antibodies. For variable temperature experiments, shifts to lower Tm values indicate a decrease in stability and higher values indicate an increase in stability. Changes in mass spectrometry indicate different structures or stoichiometries. For IM, shifts to lower CCS values indicate more compact structures while larger values indicate larger, often unfolded structures. By applying activation energy and monitoring unfolding, for example biosimilar IM, shifts in stability can be monitored by shifts in the IM peak relative to the activation energy.
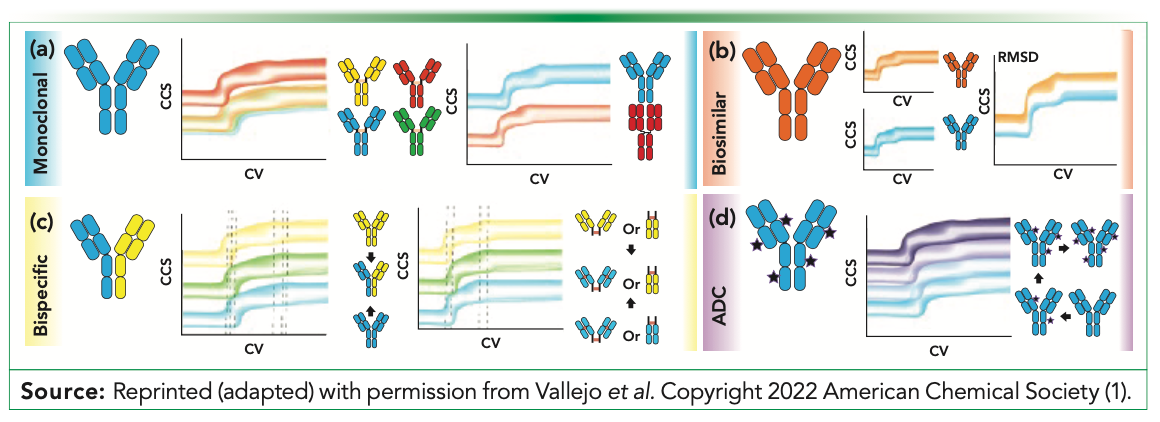
IM–MS has been shown to be a powerful tool to characterize disulfide bonding patterns, structural differences between a biosimilar and its reference product, glycosylation patterns, and several other key structural elements linked to stability. Variable temperature IM–MS can also be used to monitor structural changes where the temperature of the IM-MS drift gas is adjusted. These techniques in parallel to CIU methods can help with monitoring domain structure and anion and cation adduction and binding. In addition, Figure 2 shows representative data of how CIU can monitor disulfide bonding, glycosylation patterns, ADC drug loading, domain exchange, HDX uptake, light-chain variants, bispecific stoichiometries, and differences between reference products and biosimilars (1).

FIGURE 2: CIU applications for biotherapeutic mAbs. (a) Differentiation of monoclonal IgG subclasses by disulfide bonding patterns and difference in CIU unfolding because of domain exchange. (b) Biosimilar antibodies have qualitatively similar fingerprints, but contemporary CIU analyses can quantitate subtle differences in stability. (c) Bispecific antibodies present CIU characteristics centered between the precursor structures. (d) Shifts in CCS and stability can be quantified as a function of increasing drug load in ADC biotherapeutics.
Conclusion
Biopharmaceutical stability is a fundamental part of ensuring product quality, safety, and efficacy. It refers to the ability of a drug substance or product to retain the same properties and characteristics, within specified limits, that it had at the time of manufacture (3). Several different organizations, such as ICH, produce guidance on how to monitor drug stability throughout a product’s life cycle. The monitoring of biopharmaceutical stability requires several orthogonal techniques that include LC (for example, SEC, reversed-phase LC) and MS. LC methodologies are well established for monitoring stability and MS methodologies are becoming increasingly used. Regardless of the stability program in place, and the analytical technologies used in that program, drug stability is a cornerstone of ensuring that patients get the drugs they need when they need them.
References
(1) Vallejo, D.D.; Rojas Ramirez, C.; Parson, K.F.; Han, Y.; Gadkari, V.V.; Ruotolo, B.T. Mass Spectrometry Methods for Measuring Protein Stability. Chem. Rev. 2022, 22(8), 7690–7719. DOI: 10.1021/acs.chemrev.1c00857.
(2) Bajaj, S.; Singla, D.; Sakhuga, N. Stability Testing of Pharmaceutical Products. J. Appl. Pharm. Sci. 2012, 2(3), 129–138 DOI: 10.7324/JAPS.2012.2322
(3) Wong A.W.; Datla, A. Handbook of Pharmaceutical Analysis by HPLC, vol. 6. (Separation Science and Techonology, Mountain View, 2005), pp. 335–358.
(4) Berkowitz, S.; Rathore, A.S.; Krull, I.S. Challenges in the Determination of Protein Aggregates, Part II. LCGC North Am. 2015, 33(7), 478–489.
(5) Rathore A.S.; Krull, I.S. Challenges in the Determination of Protein Aggregates, Part I. LCGC North Am. 2015, 33(1), 42–49.
(6) Sauna, Z.E.; Immunogenicity of Protein-based Therapeutics. (accessed November 2022). https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics.
(7) International Conference on Harmonization, ICH Q1A(R2), Stability Testing of New Drug Substances and Drug Products (ICH, Geneva, Switzerland, 2003).
(8) U.S. Food and Drug Administration, Expiration Dates-Questions and Answers. (accessed November 2022). https://www.fda.gov/drugs/pharmaceutical-quality-resources/expiration-dates-questions-and-answers.
(9) World Health Organization, WHO Guidelines on Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products (accessed November 2022). https://www.who.int/publications/m/item/who-guidelines-on-stability-testing-of-active-pharmaceutical-ingredients-and-finished-pharmaceutical-products.
(10) International Conference on Harmonization, ICH Q1B, Photostability Testing of New Drug Substances and Products (ICH, Geneva, Switzerland, 1996).
(11) International Conference on Harmonization, ICH Q1C, Stability Testing for New Dosage Forms (ICH, Geneva, Switzerland, 1996).
(12) International Conference on Harmonization, ICH Q1D, Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products (ICH, Geneva, Switzerland, 2002).
(13) International Conference on Harmonization, ICH Q1E, Evaluation of Stability Data (ICH, Geneva, Switzerland, 2003).
(14) Hong, M.S.; Severson, K.A.; Jiang, M.; Lu, A.E.; Love, J.C.; Braatz, R.D. Challenges and Opportunities in Biopharmaceutical Manufacturing Control. Comput. Chem. Eng. 2018, 110, 106–114. DOI: 10.1016/j.compchemeng.2017.12.007
(15) International Conference on Harmonization, ICH Q5C, Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products (ICH, Geneva, Switzerland, 1995).
(16) Hong, P.; Koza, S.; Bouvier, E.S.P.; Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and Their Aggregates. J. Liq. Chromatogr. Relat. 2012, 30(20), 2923–2950. DOI: 10.1080/10826076.2012.743724.
(17) Brown, C.J.; Woodall, D.W.; El-Baba, T.J.; D.E. Clemmer, D.E.; Characterizing Thermal Transitions of IgG with Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30(11), 2438–2445 (2019). DOI: 10.1007/s13361-019-02292-6
ABOUT THE AUTHOR

Jared Auclair is the Vice Provost of Research Economic Development and Director of Bioinnovation at Northeastern University, in Boston, Massachusetts.

ABOUT THE COLUMN EDITOR

Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.

















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.



