Analytical Method Validation for Biotechnology Proteins, Peptides, and Antibodies
LCGC North America
The authors explore how true (complete) method validation of biopharmaceuticals must be forthcoming.
In the vast majority of previous "Validation Viewpoint" columns, we have emphasized low molecular weight (MW), conventional, synthetic, single molecule (or enantiomers) pharmaceuticals. At times, we also have devoted columns more toward biotechnology-derived products or impurities, also known as biopharmaceuticals (1–4). In one particular column installment, we discussed validation in biotechnology and well-characterized biopharmaceutical products (2), and that column should really be consulted for additional information, perhaps before the current one. Given that more and more pharmaceuticals are being derived from recombinant biotechnology proteins, especially glycoproteins, an especially difficult class of molecules to characterize fully, it would seem that guidelines and directives for true (complete) method validation of biopharmaceuticals must be forthcoming (13–14). At the moment, that which is available through ICH or FDA do not appear equivalent to the existing guidelines for synthetic, low MW pharmaceuticals (15–17). The question will then become: Why do proteins deserve a different treatment than what is now available?



We must, at the outset, indicate that characterization methods are used to characterize the drug substance product. And, that data generated by such methods are then used in regulatory filings. Typically, such assays are performed on state-of-the-art, expensive instrumentation and equipment, usually by highly trained and experienced scientists, often at the Ph.D level, within the research and development laboratories of a biotechnology firm. Such characterization studies usually are done once, perhaps twice, in the lifecycle of the final product.


Michael Swartz
As contrasted with drug substance characterization methods, other studies are required, such as impurity profiles, stability, dissolution, storage conditions, and numerous others, to make disposition decisions regarding final release of the drug product. Typically, much simpler (usually analytical) methods are performed on "commercially available" instrumentation in quality control (QC) laboratories, and usually by B.S. level personnel. Some of the following discussion addresses release–stability methods and should not be applied to characterization methods. There do not appear to be any final regulatory guidelines that address total characterization methods. Complete analytical characterization of (often) a complex mixture of species in the final, drug substance is often impossible, even given today's state-of-the-art instrumentation and methodologies (13,14). However, recently, regulators have started to discuss qualification and validation of such methods, but nothing concrete has yet appeared in the relevant literature (International Conference on Harmonization [ICH] or U.S. Food and Drug Administration [FDA]). The biopharmaceutical industry, FDA (and other regulatory agencies around the world), and ICH have long recognized the often extremely complex nature of glycoprotein biopharmaceuticals and the difficulties inherent in their complete structural characterization. This may not change in the near future (18).

Ira S. Krull
Guidelines are not requirements, but just suggestions, often made by regulatory agencies, and are always open to interpretation by the filing company. Such guidelines also are independent of the type of analytical technique or the drug substance and related drug product substances. In the biologics area of FDA (Center for Biological Evaluations and Research, CBER), which is usually where biotechnology products fall, it has become accepted practice that the process of manufacture defines the product. If the drug substance is made the same way each and every time, then it is considered by the FDA to be the very same material.
And, revalidation or recharacterization of subsequent batches is not required or desired. However, if the process is altered, it also is assumed that the product is no longer the same as before. It is likely and desirable that, in the future, more advanced characterization techniques, may lead to complete (rather than "well") characterization of complex protein products. That would help move the industry away from the current process definition of what characterizes the drug substance (namely, the process remaining the same over time).
One final subject before we actually start to discuss existing guidelines for analytical method validation of biotech products, and that has to do with something FDA calls biologics licensing application (BLA), which is not the same as an investigational new drug (IND) application to FDA. INDs are used for small molecule, low molecular weight (MW) drug substances, but BLAs are used for biotechnology products, in most cases, though some biotech products (for example, insulin) are so simple that they used INDs. As it now seems that most biotech products of the future will be considerably more complex than insulin, we would expect that the use of BLAs will remain for quite some time, within CBER/FDA. Usually, after extensive clinical testing is completed successfully, a New Drug Application (NDA) must be filed before marketability with the FDA. For biogenerics or follow-on biologics, being introduced by a generics firm, an Abbreviated New Drug Application (ANDA) must be filed, often without extensive clinical or biological testing, if true and complete chemical equivalency can be demonstrated fully and satisfactorily. This is often a tricky matter.
Current Guidelines for Analytical Method Validation of Biotechnology-Derived Pharmaceuticals
In an informal survey of some (not most) biotech firms, it would appear that, at present, there are no specific guidelines (ICH, EMEA, or FDA) on validation methods for biotechnology products, especially proteins (5). Apparently, the industry follows ICH Q2R1 and Q6B (6,7,15,16). In 2000, the FDA issued a Guidance for Industry entitled "Analytical Procedures and Method Validation," but this document references ICH Q2 as the source document, so it is not really different (7). Some biotechnology people incorrectly apply FDA's "Guidance for Industry — Bioanalytical Method Validation," for the validation of bio- and immuno-assays, to protein products (18). This document has been designed to be applied to bioanalytical methods used for nonhuman, pharmacology, and toxicology and preclinical studies. Bioanalytical method validation is not the same thing as method validation for biotechnology-derived products, as they are two totally different areas. Bioanalytical method validation really applies, in general, to the analysis of synthetic drugs in biological samples! It also could be applied to the analysis of larger, biopolymer-derived drugs (biotechnology drugs or biopharmaceuticals), also in biological type samples. However, those guidelines do not apply to biotechnology drug method validation, per se!
In 1999, ICH issued a guideline entitled "Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, ICH Harmonised Tripartite Guidelines, Q6B, Step 4" (8,15). If one reads through this document, it is apparent that it has little specifically to do with analytical method validation for protein-derived biopharmaceutical products (2.2.2 Validation of analytical procedures). That is, it really refers to other ICH validation guidelines, originally put forward for low MW pharmaceuticals but not specifically for biotechnology-derived ones. There are at least two other, related reviews on the analytical lifestyle of biotechnology products that should be referenced (9,10). These are very good overviews of how analytical validation requirements can change during the lifecycle of a biotech product and why.
We and others believe that proteins and their analytical methods deserve a different treatment for validation than what is being used currently, which were originally designed and approved for low MW, synthetic pharmaceuticals. It is also the case that ICH Q2 does not really meet the current needs of the biotechnology industry. And, given that biogenerics are being introduced at a very rapid pace outside of the U.S., one would expect this to occur here in the U.S. in the near future (11). ICH and FDA need to address what guidelines and requirements will be issued to assist the biogeneric firms, obviously eager to introduce biogenerics to the U.S. marketplace.
We hasten to add that the industry and FDA both have come to agree that many classes of protein biopharmaceuticals are so complex that they cannot, at present, be fully characterized (12). This conclusion has led to the euphemistic term "well characterized biopharmaceutical products," as opposed, it is assumed, to the term "fully characterized biopharmaceutical products." As described on the California Separations Society (CaSSS) website, organizers for the Well Characterized Biopharmaceutical Products (WCBP) series of meetings, such meetings are designed to be at the interface of regulatory and analytical sciences for biotechnology health products. In part, as a result of industry and FDA having such meetings, as well as others, a consensus has arisen that as biotechnology-derived protein products may indeed never be fully characerizable, method validation guidelines should be different than those for low MW pharmaceuticals. And this is precisely what has now evolved over more than a decade. The question could be raised, has this been a good or a bad thing for the industry and public? And, will the current situation ever evolve and improve, if that is the right word, in terms of providing guidelines that truly meet the needs of the biotech industry and the public?
Biotechnology Versus Synthetic Drug Products
For those involved with the biotech industry, it is obvious that the major difference between these two groups of synthetic, small molecule drugs and high MW, complex proteinaceous species has to do with molecular weight, conformations possible, solubility, stability, lifetimes in vivo, posttranslational modifications, and microheterogeneity (19,20). In general, with low MW drugs, the chromatographic or electrophoretic peaks are single species, perhaps enantiomers or diastereomers, but are fairly easy to purify and identify. With many, if not most, protein-derived biopharmaceuticals (especially complex glycoproteins), other than for low MW, unmodified peptides or polypeptides, quite the opposite is usually the case. That is, high MW glycoproteins and antibodies often have high degrees of microheterogeneity, numerous different isoforms or posttranslational modifications (PTM), and different conformations of the very same species, leading to broad, often ragged chromatographic or electrophoretic peaks (13,14). The appearance of trains or trails of spots in two-dimensional gel electrophoresis (2DGE) is often indicative of families of PTMs, often glyco- or phospho-derived. These often are unresolvable and perhaps impossible to resolve and then identify using any current analytical methodologies (13,14,19,20).
There is something called "dynamic microheterogeneity" such as the extent of oxidation of methionine (Met), wherein sulfoxide and sulfone forms can form to varying degrees within a protein and from site to site of that amino acid. The presence of numerous variants or isoforms of the drug substance can further lead to a broadening and shifting of some peaks in a chromatogram, which only leads to more concern on the parts of both the industry and FDA. The presence of numerous isoforms, the same basic protein but having different isoelectric points (pIs), leads to the presence of numerous variants, but these are (again) all part of the final drug product (as in the case of glycoproteins) mentioned previously.
Even mass spectrometry (MS) is often unable to resolve and fully characterize any or all of the species present under a single chromatographic or electrophoretic peak or band or spot. Even multidimensional separations techniques, involving several high performance liquid chromatography (HPLC) or gel electrophoresis steps, using orthogonal separation modes with MS, often are unable to fully resolve all the variants present for very complex glycoprotein products. Other than for low MW, unmodified (no PTMs) peptides or polypeptides such as insulin or human growth hormone, biotechnology is not dealing with single molecular entities. In perhaps too many instances, the absolute identity or structures of these species is inferred, not fully proven. And, in the absence of a fully characterized protein product, it is then impossible to obtain an authentic reference standard as a single entity, or to perform true, absolute quantitation! Such a situation is a very important point, for the ability to perform absolute quantitation lies at the very heart of many of today's ICH guidelines for complete analytical method validation of low MW, synthetic drugs. That should then lead to the realization that the current ICH guidelines for such (low MW) synthetic drugs cannot be applied fully or directly to most biotech-derived biopharmaceutical (protein) products. There is a misfit somewhere in trying to make a round peg fit into a square hole, and that is basically what has evolved in attempting to relyupon ICH guidelines for method validation of synthetic, low MW drugs, and directly applying them to high MW, protein type products.
We realize that there are and will be other classes of biotech derived products introduced to the marketplace. These have been and will be classes such as carbohydrates, lipopolysaccharides, synthetic RNA derivatives, polynucleotides, DNA-derived species, phospholipids, and others. There is nothing obvious in the ICH guidelines aimed at such species, as yet, and one might assume that those now being used for proteins also will be applied for these other classes. However, which aspects of existing ICH guidelines for method validation of low MW drugs should be assumed applicable to biotech proteins is very unclear in anything to be found online, at either ICH or FDA websites.
It almost appears that the industry intuitively knows what FDA expects or wants to see in filings for newer and generic protein products. And, such filings can vary from company-to-company, product-to-product, as the industry believes is needed and expected of their newest protein product going to FDA for approvals. The current guidelines do not spell out fully what extent of analytical characterization and method validation is going to be accepted and approved for an NDA or ANDA. Some degree of communication between the filing company and the regulatory agencies, before filings, is recommended to avoid later misunderstandings and confusions.
What Are the Current ICH Guidelines for High MW Biopharmaceuticals Being Produced and Marketed Today?
Though we have tried to locate specific guidelines on both the ICH and FDA websites for biopharmaceutical proteins, they do not really appear to exist as clearly as for low MW drugs. Yes, there are documents such as Q6B and Q2(R1), but these were developed originally for low MW drugs [Q2(R1)] or they do not describe true validation guidelines (Q6B and others in this genre). There is no similar set of guidelines for proteinaceous pharmaceuticals to compare with Q2(R1), but that is what the industry currently uses, with some serious changes. That is, in Q2(R1), there are found the usual guideline steps, stage dependent- accuracy, precision (intermediate precision, reproducibility), limits of detection (LOD), limits of quantitation (LOQ), specificity, linearity, range of linearity, system suitability, and robustness. However, several of these processes are often impossible to pursue with complex and complicated glycoproteins, as well as for other mixtures of phosphoproteins and any protein with a high number of variants or isoforms. Often, there are so many variants in the drug substances, especially for glycoproteins, that it is just not possible to qualitatively identify, with a high degree of specificity, the exact structures of such variants (13,14). This then means that usually it is not possible to have authentic reference materials with which to perform absolute protein quantitations, as is done routinely for low MW drugs. And, if absolute quantitation is not feasible, then there is no way to perform accuracy or precision of such quantitations. And that also means it is not possible to derive LOD or LOQ in terms of absolute mass or concentrations. Then there is not a need to derive calibration plots, linearity equations for such plots, or ranges of linearity, and so forth. This could greatly reduce the total number of steps needed to perform possible method validation for complex biopharmaceuticals. And that is precisely what has evolved, and why we have well characterized biopharmaceutical products, but not fully characterized or validated ones today, as we do for low MW synthetic drug substances.
While it is true that smaller peptides, containing but one variant and no isoforms, can be quantitated in an absolute sense, using external standards (synthetic), as well as calibration plots, linearity, accuracy and precision in spiked samples, this cannot generally be done for more complex proteins. With such low MW drugs, one can demonstrate true accuracy and precision in real samples, as well as intermediate precision and repeatability, method transfer, system suitability tests, and robustness. However, such complete validation packages are no longer feasible with high MW polypeptides or proteins when several variants are possible, such as with complex glycoproteins and antibodies.


Figure 1: The percentages of antibody C-terminal lysine isoforms quantitated by peak areas on both cation-exchange HPLC and cIEF assays (n = 8, y-error bar is SD) (21,22).
What has evolved are more "qualitative" methods of quantitation for protein drug substances, relative percent peak areas of all variants present, and a demonstration that these ratios are constant from batch-to-batch, through stability studies and so forth (Figures 1 and 2). Figure 1 illustrates the relative percentages present for C-terminal lysine (Lys) variants (isoforms) of a human, monoclonal antibody against tumor necrosis factor (TNF-alpha) (21,22). These have been "quantitated" using both cation-exchange HPLC and capillary isoelectric focusing (cIEF) type assays. Figure 2 illustrates the linearity of the cation-exchange HPLC assay for these same antibody variants, containing 0-, 1-, or 2 C-terminal Lys present. Figure 2 contains a plot of peak area (y-axis) versus injection amount (micrograms) for each of the three variants, along with the equations of their straight lines and correlation coefficients (21,22). It is not a plot of the absolute amounts of each variant versus peak areas, but of injection amounts of total antibody variants. The absolute amounts of each variant could be derived from the relative peak areas times the total injection amount, assuming that each variant has the same UV absorbance intensity in the cation exchange–UV assay. It often is very difficult to obtain true, separate standards for each such variant, and here there are only about three such variants for the antibody. What happens when there are 10 or 20 such variants?

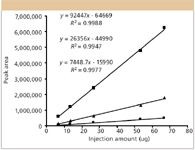
Figure 2: Linearity of the cation-exchange HPLC assay. The calibration curves are O-Lys, 1-Lys, and 2-Lys of the antibody from top to bottom, with equations of the straight lines and correlation coefficients, as indicated (21,22)
Because there is no need to worry about low levels of protein variants (these are not impurities) in the drug substance, in most cases, LOD and LOQ also are unwarranted validation steps. Remember, true variants of one another are a part of the final drug substance and product. What is needed is a clear, valid demonstration of chemical equivalency, batch-to-batch and thru all stability studies. Given that many complex mixtures of glycoproteins cannot, at present, be fully characterized, gross characteristics of the drug substance are demonstrated, such as total glyco contents, individual glyco components or patterns, and when possible, exact location of glycos and protein primary sequence with all PTMs denoted. Impurities also must be identified and quantified in terms of relative percent peak areas, just as they would be for low MW drugs (13,14,19,20).
Figure 3 illustrates the use of high performance capillary electrophoresis (HPCE) to characterize the glycoform content of two glycoprotein samples (Sample A and Sample B), applied to intact glycoforms, as antennary species (13–14,23). Similar HPCE approaches have long been used to characterize individual monosaccharides, after the branched–antennary polysaccharides have first been reduced to their monosaccharide components (23). This is easy to do if one is dealing with a single, glycoprotein entity, but if there are numerous such species together or there are numerous sites of glycosylation, then this (and other) techniques cannot fully characterize such glycoproteins after the glycoforms are removed from the protein backbones. There are, of course, other, usually MS approaches, that also can be used successfully to characterize glycoforms and glycos derived from intact glycoproteins or even the intact glycopeptides without first releasing the glycos (13,14,19,20).


Figure 3: Carbohydrate labeling and analysis kit, microheterogeneity determination. Quantitation of relative oligosaccharide concentrations. (PA800 Protein Characterization, Beckman Application Note, 2009, Beckman-Coulter Corporation, Fullerton, California).
What Has Been Changed by ICH and FDA in Their Requirements for High MW Biopharmaceuticals?
So, then what is really left in the way of method validation for biopharmaceutical proteins? Specificity is still possible to demonstrate, by providing HPLC or HPCE or MS methods that can resolve the true drug substances from other species perhaps present, such as impurities or excipients, as in Figure 4. This figure represents the inherent ability, in this instance, of cation exchange to fully resolve simple variants in the drug substance, differing by a single amino acid at the C-terminal. However, the broadness of each peak suggests the possible presence of various glycoforms as also being present on each variant (21,22). And, individual variants can be identified, often by combinations of HPLC–MS or HPCE–MS or other common analytical techniques (for example, 1-D gel electrophoresis), and shown to be consistent batch-to-batch. Robustness also can be demonstrated, or how small, unintentional changes in one or more operational parameters might change, for example, the peak shape in HPLC, resolutions, plate counts, peak symmetry, and even relative percent peak areas; or how small operational changes in the MS might affect relative peak areas in a total ion chromatogram for the total drug substance; or, how small changes in the operations of the MS might change the ability to fully characterize or quantitate an individual protein variant in the drug substance. System suitability testing is still possible with biopharmaceuticals, especially if individual protein variants are available for demonstrating performance of the HPLC, HPCE, or MS instrumentation and validated methods. For example, if conversion of asparagine to isoaspartic or aspartic acids has occurred somewhere within a protein, then a system suitability method would have to show resolution of these species by HPLC or HPCE (13,14,19,20). Such methods already exist but are rarely, if ever, required by FDA or suggested by ICH. Such methods require showing that the drug substance peaks are fully baseline resolved from one another, when possible, under the validated operational conditions for the method. System suitability has not yet become a required step in method validation for low MW drugs, as it is only in a guideline. System suitability is not in the USP guidelines, it is treated in a separate USP chapter. Note that we also are addressing areas such as stability studies and cleaning validation in this column, because methods for performing these also must be validated for biopharmaceuticals.

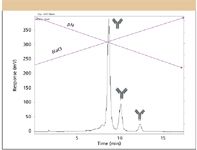
Figure 4: Analysis of antibody 1 by cation exchange. The chromatogram is the intact Ab1 with neutral oligosaccharide peaks being (left to right) C-terminal O-Lys, 1-Lys, and 2-Lys, with structure indicated above each peak. The front shoulder peak of the main peak is an acidic peak (21,22).
What Might Be Required in the Future by ICH for High MW Biopharmaceuticals, Regarding Analytical Method Validation?
To some extent, ICH and FDA have tried to come to terms with the often extreme complexity of many biotechnologically-derived biopharmaceutical drugs. This was really the reason for establishing the entire WCBP series of meetings for the past 12 years or so. However, ICH has not been extensively involved in such meetings, though FDA has been a significant presence over the years. Both of these groups have come to realize that it makes no sense to require analytical method validation steps that cannot be realized readily by the
industry for most protein products. FDA does not want to establish guidelines or requirements that cannot be met, readily or at all, by the industry, which would only keep potentially lifesaving drugs from going to market. Biological efficacy and safety may trump any (potentially impossible) demonstration of complete structure determinations of all variants present in a complex mixture of glycoproteins. If FDA or ICH sets up a series of guidelines or requirements that cannot be met for most biopharmaceuticals, that is the end of most of the biotech industry. It is nice to suggest that we require all biotech firms to demonstrate absolute quantities, accuracy and precision, as well as LOD and LOQ for all variants in a complex mixture of glycoproteins, or other protein products, but that is somewhat naïve and self-defeating. Again, the issue is biological–clinical efficacy and patient safety, as well as the ability to demonstrate chemical equivalency batch-to-batch for a given product.
However, what about biogenerics then? Will the FDA require more of these firms for introduction of their products than for the original, proprietary manufacturers? Will they now be made to demonstrate true chemical equivalency in terms of absolute structures of all species present, even if these were never identified fully in the proprietary products years ago? And, to also demonstrate for the generics absolute levels of each variant, rather than relative percent peak areas in HPLC or HPCE? Will the biogenerics be made to repeat all the same clinical studies originally demonstrated by the proprietaries, as well as true chemical equivalencies? Where does the game end then? Is the goal to encourage biogenerics coming to market or to prevent this? Will the method validation requirements for biogenerics be identical to those for proprietary biopharmaceuticals or not? How will they differ, if they do differ, and why? These are very thorny and tricky questions that ICH and FDA are now trying to sort out (11). But, at the moment, it is not at all clear just where FDA will come down on the issue and what they will or will not require of biogenerics. The recent Biogenerics 08 meeting in Boston addressed these very questions, with a number of FDA speakers addressing these and other questions (11). However, at the moment, there are no true method validation guidelines for biogeneric biopharmaceuticals, and it is unknown just when those might appear, if anywhere. This is clearly a very difficult issue for FDA to resolve, and it is not yet clear if ICH is addressing it in full. Only time will tell.
Conclusions
We have tried to address the important issue of where do analytical method validation guidelines and directives stand today for biopharmaceutical protein products. What seems to have evolved are a different set of, perhaps unstated, guidelines and accepted practices by FDA, for acceptable submittals of "validated" analytical methods in INDs, NDAs, ANDAs, and so forth. Obviously, the meaning of the word "validated" differs between low and high MW drug products, and FDA has come to accept that what is expected of all low MW, single molecule entities (even enantiomers), cannot be asked of complex mixtures of glycoproteins or other complex mixtures of protein biopharmaceuticals. Perhaps this is as it should be and more cannot be required or expected at present. What happens, though, when the analytical methods evolve to such an improved level that it does become possible to fully characterize all glycoproteins present in a complex mixture? What happens if it is then possible to synthesize most or all of the major components of a mixture and demonstrate true quantitation, accuracy, precision, LOD, LOQ, calibration plots, and linearities, thereof? What will FDA or ICH then suggest be changed in the current guidelines, white papers, directives, and expectations in new submittals? Anything? Nothing? Again, only time will tell.
Michael Swartz "Validation Viewpoint" Co-Editor Michael E. Swartz is Research Director at Synomics PharmaceuticalServices, Wareham, Massachusetts, and a member of LCGC's editorial advisory board.
Ira S. Krull "Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor ofchemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.
References
(1) M. Swartz and I.S. Krull, LCGC 25(8), 718–724 (2007).
(2) I.S. Krull and M.E. Swartz, The Pharmaceutical Regulatory Guidance Book, 18-23 (July 2006).
(3) M.E. Swartz and I.S. Krull, LCGC 25(12), 1196–1201 (2007).
(4) I.S. Krull, P.T. Kissinger, and M. Swartz, LCGC 26(11), 110–1117 (2008).
(5) I. Apostol. Private communication (January 2009).
(6) International Conference on Harmonization (ICH), website: www.ich.org.
(7) US Food and Drug Administration, Guidance for Industry-Analytical Procedures and Method Validation, 2000. www.fda.gov.
(8) ICH Harmonised Tripartite Guideline, Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, March, 1999. www.ich.org.
(9) I. Apostol and D.N. Kelner, BioProcess Int., 12–20 (2008).
(10) I. Apostol and D. Kelner. Managing the analytical life-cycle for biotechnology products. A journal from method development to validation. Part Two. 12-18 (2008).
(11) Biogenerics 2008, Meeting held in Boston, Massachusetts, Spring, 2008, The Barnett Institute at Northeastern University.
(12) California Separations Society (CaSSS), Well Characterized Biopharmaceutical Products annual meetings, San Francisco, California, 1998–2009, www.casss.org.
(13) Post-translational Modifications of Proteins. Tools for Functional Proteomics, Second Edition, C. Kannicht, Ed. (Humana Press, Totowa, New Jersey, 2008).
(14) R. Westermeier, T. Naven, and H.-R. Hopker. Proteomics in Practice. A Guide to Successful Experimental Design, Second Edition (Wiley-VCH, Weinheim, Germany, 2008).
(15) ICH Harmonised Tripartite Guideline, Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, Q6B, Current Step 4 Version, Dated 10 March, 1999, International Conference on Harmonisation, Geneva, Switzerland, www.ich.org.
(16) ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1), Current Step 4 Version, Parent Guideline on Methodology, dated 6 November 1996, incorporated in November 2005, International Conference on Harmonisation, Geneva, Switzerland, www.ich.org.
(17) Guidance for Industry, Bioanalytical Method Validation, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine (CVM), Rockville, Maryland, May 2001.
(18) MSB 09 Meeting, Boston, Massachusetts, February 2009, Microseparations in Biology, California Separations Society Meeting (CaSSS), www.casss.org.
(19) R. Kellner, F. Lottspeich, and H.E. Meyer, Microcharacterization of Proteins, Second Edition (Wiley-ICH, Weinheim, Germany, 1999).
(20) Emerging Technologies in Protein and Genomic Material Analysis, G. Marko-Varga and P. Oroszlan, Eds. (Elsevier Science Publishers, Amsterdam, 2003).
(21) L.C. Santora, Ph.D. dissertation, Characterization of recombinant, human, monoclonal antibody using CIEX, SEC, and MS. Northeastern University, Boston, Massachusetts, January 2001.
(22) L.C. Santora, I.S. Krull, and K. Grant, Anal. Biochem. 275, 98 (1999).
(23) Carbohydrate Labeling and Analysis, PA800 Protein Characterization, Beckman Application Note, 2009, Beckman-CoulterCorporation, Fullerton, California.

")













, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




