Advances in High-Speed and High-Resolution Ion Chromatography
Special Issues
The speed of IC has been limited by pressure and flow limits of the hardware. Recent advances are enabling faster IC separations.
Compared to modern high performance liquid chromatography (HPLC) and ultrahigh-pressure liquid chromatography (UHPLC), separation speeds in ion chromatography (IC) have lagged behind. In recent years, dramatic strides have been made by the development of PEEK flow components, eluent generators and membrane suppressors with increased pressure and lower volumes, smaller particle size packings that permit shorter columns and higher flow rates, and capillary columns with higher efficiencies. Here, we review these developments and show examples of faster IC separations. For simple systems, separation speeds rival those of modern LC. For complex samples, separation times are still slow, but the use of 2D techniques allows more rapid analysis of complex samples in high ionic-strength matrices.
Just four decades ago, the analysis of an individual ion required that the analyst perform a time-consuming titration or gravimetric measurement. Each additional ion to be analyzed magnified the effort required. In 1975, Small and coworkers (1) introduced ion chromatography (IC). This technique enabled the simultaneous analysis of multiple ions with low detection limits. Compared to the hours of labor needed for classical methods, IC yielded rapid measurements. For instance, Li+ , Na+ , and K+ could be baseline-resolved in 134 min (2). Similarly common anions could be analyzed in 20 min (1,2).
Today, IC is widely used for the analysis of inorganic ions and many ionizable organic compounds. More than 70 commercial IC columns are available (3). Until recently, however, developments by IC column manufacturers focused on increasing the ion-exchange capacity, reliability, and selectivity of stationary phases (4) rather than the speed of analysis. Thus, up to just a few years ago the typical separation time of anions differed little from that demonstrated by Small and coworkers in 1975 (1,2)
In contrast, in the last decade, rapid developments have taken place in reversed-phase liquid chromatography. Smaller particles have enabled faster analysis by allowing shorter column lengths while maintaining high efficiency and resolution (5). Introduction of sub-2-µm particles required ultrahigh-pressure (up to 15,000 psi) metallic pumps because of the dramatically greater back pressure generated by these smaller particle diameters (5,6).
Why has IC lagged behind this trend? First, IC was limited by the hardware (7). The stainless steel pumps, tubing, and fittings used in high performance and ultrahigh-pressure liquid chromatography (HPLC and UHPLC) are not compatible with the highly corrosive acidic or alkaline eluents required in IC. Early IC systems were constructed with glass columns and polyoxymethylene (Delrin) components (8). The introduction of polyether ether ketone (PEEK) pumps and tubing increased the pressure capabilities of IC to near that of conventional HPLC systems. Second, severe baseline drift and ghost peaks were experienced with gradient elution in IC because of the inherent background conductivity of carbonate eluents and impurities in the carbonate or hydroxide eluents. The generation of ultrahigh-purity eluents such as carbonate-free sodium hydroxide was made possible through membrane-based electrodialytic eluent generators (9,10). The fragility of the eluent generators restricted the upper pressure limit to 3000 psi (11). The maximum flow rate in IC was further limited by the suppressor to 1–3 mL/min (for 2 mm to 4 mm columns, respectively) to protect the membranes from leaking. Thus the standard IC hardware was pressure- and flow rate–limited as late as 2011.
Because of these pressure and flow rate limits, IC columns continued to be characterized by large particle sizes (7–13 µm) and long (20–25 cm) columns. For instance, Figure 1a shows a typical ion chromatogram using a 25-cm IonPac AS22 column packed with 6.5-µm particles. Seven common anions are separated in ~12 min with 9600 plates at a column pressure of <2000 psi.



Figure 1: Comparison of separations obtained using 25- and 15-cm columns with the same particle size (6.5 µm) and column diameter: (a) 25 cm à 0.4 cm AS22 (flow rate: 1.2 mL/min), (b) 15 cm à 0.4 cm AS22-Fast (flow rate 1.2 mL/min), (c) 15 cm à 0.4 cm AS22-Fast (2.0 mL/min). The typical working pressure of AS22 and AS22-Fast is ~1900 psi. The excessive resolution is shown in blue boxes. Eluent: 4.5 mM sodium carbonate, 1.4 mM sodium bicarbonate; injection volume: 10 µL; suppressed conductivity detection. (Courtesy of Thermo Fisher Scientific.)
However, there is actually excessive resolution in Figure 1a, leading to "wasted" time, which is highlighted in blue in the figure. Clearly, some of this excess resolution could be traded for analysis time by shortening the column while keeping the particle size and other parameters the same. The first such column was the 15-cm IonPac AS22-Fast column, which was introduced at Pittcon 2010. Figure 1b shows a 7-min separation on the AS22-Fast column — a 40% reduction in run time. The efficiency is reduced from 9600 to 5000 because of the shorter column length, but it is still sufficient for baseline resolution. The lower back pressure of the shorter column also allows use of higher flow rates while staying within the pressure limits of the IC instrumentation. Figure 1c shows a 4.5-min separation of seven common inorganic anions using a 15-cm AS22-Fast at 2 mL/min. Using even shorter columns while keeping the particle size the same can further reduce the analysis time. For instance, Haddad's group used 3–5 cm columns packed with 7.5-µm resins to demonstrate 15-min separations of highly retained perchlorate and thiocyanate ions (12). Such ions are typically retained for more than 1 h on standard 25-cm IC columns.
The next evolution in the speed of IC analysis was associated with the redesign of the commercial hardware for capillary columns (~0.4 mm i.d.). Such columns allow the same linear velocity (u, cm/min) as a normal 4-mm i.d. column at a 100-fold lower volumetric flow rate (13), enabling extended operation with a single bottle of eluent. An immediate consequence of the lower volumetric flow rates needed for capillary IC was a decrease in the surface area of the electrodialytic membrane in the eluent generator (14,15), which yielded a higher burst pressure for the membrane. Thus, a capillary IC system such as the Thermo Scientific Dionex ICS-5000 has a pressure limit of 5000 psi (compared to a limit of 3000 psi for a conventional IC system). Capillary separation systems also produce greater column efficiencies (reduced plate heights <2) than large-bore counterparts (13). Figure 2 shows the separation of seven common anions in less than 2 min using a 15-cm capillary column packed with 4-µm particles (16). At 25 µL/min, the back pressure was only 3480 psi. For the sake of comparison, this capillary separation in Figure 2 takes 17 s/ion compared to 98 s/ion for the AS22-Fast column in Figure 1c. Thus, capillary systems have the potential of high-speed IC separations by employing smaller particles in 15-cm capillaries. A number of such "fast" IC capillaries are commercially available for cations and anions. In January 2013, a redesign of the eluent generators for conventional flow IC yielded the Thermo Scientific Dionex ICS-5000+ system, which also has a 5000-psi pressure limit (17).

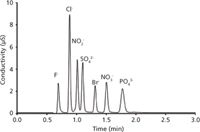
Figure 2: Fast separation of seven common anions on an IonPac AS18 capillary column (150 mm à 0.4 mm) packed with 4-µm particles. Eluent: 35 mM OH- with a flow rate of 25 µL/min; operating pressure = 3480 psi. Suppressed conductivity detection. Adapted from reference 16.
Given the ubiquitous presence of UHPLC systems in the industry, chromatographers have begun to ask "Can IC be as fast as UHPLC?" Although the terms "fast" and "ultrafast" separations are relative, a survey of the literature and commercial catalogs shows that these terms imply subminute separations. Subminute IC separations were pioneered by the groups of Paull (18) and Lucy (19). For instance, Figure 3 shows the ultrafast separation of seven common anions in just 40 s using an ultrashort 1.3-cm column packed with 1.8-µm C18 silica particles (20). Although significant developments have taken place in achieving fast separations in commercial IC columns, we have yet to see subminute IC separations in commercial columns. Technology exists to synthesize 2-µm highly crosslinked polymer particles that can withstand pressures of >5000 psi (21). However, there are still significant challenges in using smaller particles. First, as columns become more efficient, extracolumn band broadening effects come into play (20), and those effects are particularly challenging in IC where postcolumn devices such as suppressors are essential. Second, optimum packing of sub-2-µm particles is not a trivial task. Even 4.4-µm charged polymeric particles display unexpected behavior in the packing process (22). Third, the mechanical strength of PEEK limits the pressure of current IC systems to <5000 psi. IC certainly has room to catch up with the efficiencies and speed of UHPLC.

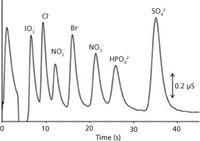
Figure 3: Ultrafast separation of seven common anions on very short column (1.3 cm à 0.46 cm) with suppressed conductivity detection. Column: Cationic surfactant (didodecyldimethylammonium bromide) coated Extend-C18 column; flow rate: 2.0 mL/min; particle size: 1.8 µm; eluent: 2.5 mM 4-hydroxybenzoic acid (pH 10.1). Adapted from reference 20.
In the above discussion, we provided examples of fast separations of a few ions in relatively simple samples by employing smaller particles or very short columns, or both. Speed becomes a secondary factor, however, when analyzing difficult and ill-characterized samples such as food, biological samples, or environmental waste. In these cases, the target is to fully resolve all the ions of interest. The factors that affect the resolution (R) of critical pair of analytes in a given separation are described by

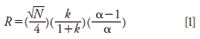
The plate number, N, is easily increased by using longer columns or smaller particles. The selectivity factor, α, and retention factor, k, become important for high resolution. Both of these variables are dependent on the stationary phase chemistry and the eluent (4,23). Software packages for the simulation and optimization of separations are available (24). For instance, the Virtual Column Separation Simulator (Thermo Fisher Scientific) uses a database of experimental retention data to predict retention using the linear solvent strength model–empirical approach. Resolution maps are also generated by the software for a critical pair of ions. Retention times have been predicted within 3% of the observed behavior for 33 anionic and cationic analytes across a variety of column formats and complex gradients (25).
Thus, by using specially designed high capacity columns and small particles, one can achieve very high resolution. Figure 4 illustrates a high-resolution separation of 44 inorganic and organic ions in 40 min using a long 25-cm capillary and 4-µm particles. This corresponds to a separation speed of 53 s/ion.

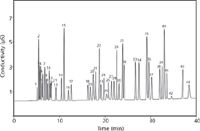
Figure 4: High-resolution gradient separation of 44 anions on a specially tailored high-capacity capillary column (prototype IonPac AS11-HC, 250 mm à 0.4 mm, 4-µm particle size). Eluent: KOH: 1â14 mM KOH in 16 min, 14â55 mM KOH in 24 min, 15 µL/min. Injection volume: 0.4 µL. Peaks: 1 = quinate, 2 = fluoride, 3 = lactate, 4 = acetate, 5 = 2-hydroxybutyrate, 6 = propionate, 7 = formate, 8 = butyrate, 9 = 2-hydroxyvalerate, 10 = pyruvate, 11 = isovalerate, 12 = chlorite, 13 = valerate, 14 = bromate, 15 = chloride, 16 = 2-oxovalerate, 17 = nitrite, 18 = ethylphosphonate, 19 = trifluoroacetate, 20 = azide, 21 = bromide, 22 = nitrate, 23 = citramalate, 24 = malate, 25 = carbonate, 26 = malonate, 27 = citraconitate, 28 = maleate, 29 = sulfate, 30 = alpha-ketoglutarate, 31 = oxalate, 32 = fumarate, 33 = oxaloacetate 34 = tungstate, 35 = molybdate, 36 = phosphate, 37 = phthalate, 38 = arsenate, 39 = citrate, 40 = chromate, 41 = iso-citrate, 42 = cis-aconitate, 43 = trans-aconitate, 44 = iodide. (Courtesy of Thermo Fisher Scientific.)
However, in most real samples not all ions are in similar concentration. For example, bromate is a human carcinogen whose concentration is regulated by various government environmental agencies around the world (26). In IC, bromate is eluted early, close to the chloride peak. In clean, low ionic strength samples, baseline resolution is satisfactory, as shown in the lower trace in Figure 5. In high-ionic-strength water samples, such as seawater or wastewater, the resolution of bromate with the adjacent peak is severely compromised, as shown in the upper trace of Figure 5 (27). The high concentration of the matrix ions overloads the analytical column. The resulting wide fronted or tailed peak broadens and shifts the retention time of the trace (~10 ppb) bromate peak (28). Eventually, the broadening compromises accurate quantification and degrades the resolution of bromate from the overloaded peak.

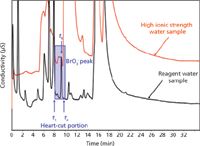
Figure 5: First-dimension analysis of a bromate peak in reagent water and high ionic strength water. The bromate concentration is 15 µg/L. The bromate retention time (tR) is 9.2 min. The start of the cut window (t1) is 8.0 min and the end of the cut window (t2) is 9.8 min. Column: 25 cm à 0.4 cm IonPac AS19; eluent: 10 mM KOH, step changed to 65 mM KOH following the elution of bromate at 1.0 mL/min; injection volume: 1.0 mL. (Courtesy of Thermo Fisher Scientific.)
One ingenious way to obtain high-resolution separation in complex matrices is matrix elimination IC (27,29,30) based on high-resolution two-dimensional IC (2D IC). In this heart-cutting approach for bromate analysis (EPA Method 302.1), the bromate ion is separated in the first dimension using a high-capacity column capable of handling the high-ionic-strength matrix. A ~2 mL portion of the suppressed eluent (the heart-cut portion) containing the bromate (t1 to t2 in Figure 5) is passed through a concentrator column positioned in place of the sample loop of the injection valve for the second-dimension separation. The trapped analyte is eluted off the concentrator column into a second-dimension IC column of different selectivity than the first-dimension column. For high-resolution applications, a second-dimension column with a lower cross-sectional area is used to enhance sensitivity (31). Figure 6 shows the second-dimension high-resolution separation of trace levels (15 ppb) of bromate in both reagent and a high-ionic-strength water sample. This heart-cutting matrix elimination approach is generic and can easily be applied on IC samples that give poor resolution of trace components as a result of matrix overload. For instance, perchlorate has similarly been resolved in highly complex water samples (31). Research is ongoing on comprehensive 2D IC for complete analysis of complex ionic samples (32).
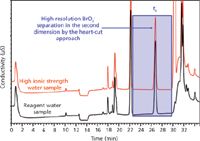
Figure 6: High-resolution separation of bromate ion in second-dimensional analysis of 15 µg/L bromate in reagent water and high ionic water matrix. Column: lonPac AS24 analytical column (25 cm à 0.2 cm) and AG24 guard column (5 cm à 0.2 cm) were used for the second dimension analysis; eluent: isocratic 10 mM KOH changed to 65 mM KOH following the elution of bromate for approximately 10 min and re-equilibrate at 10 mM hydroxide before injection; eluent flow: 0.25 mL/min. (Courtesy of Thermo Fisher Scientific.)
Conclusions
Until recently, the speed of IC was limited by the pressure and flow rate limits of the IC hardware. These conditions required the use of large particles and long columns. Numerous recent advances have enabled faster IC separations by reducing the column length while using large particles, or by reducing the particle size to 4 µm. However, commercial IC has not yet achieved the ultrafast subminute separations that have been demonstrated by sub-2-µm particles in very short columns. Thus, further advances in IC speed for simple samples are anticipated.
For complex matrices, the enhanced pressure capabilities of modern IC are enabling greater one-dimensional peak capacities. Commercial equipment has made targeted two-dimensional IC separations routine.
Acknowledgments
The authors are grateful to Chris Pohl, Kannan Srinivasan, and Rong Lin (Dionex, Thermo Fisher Scientific, California) for providing data for this review. Funding from Natural Sciences and Engineering Research Council of Canada (NSERC) and Thermo Fisher Scientific is gratefully acknowledged.
References
(1) H. Small, T.S. Stevens, and W.C. Bauman, Anal. Chem. 47, 1801–1809 (1975).
(2) H. Small, J. Chem. Educ. 81, 1277–1284 (2004).
(3) Thermo Fisher website (http://www.dionex.com/en-us/products/columns/ic-rfic/lp-71774.html) and the Metrohm website (http://www.metrohm.com/com/Produkte2/IC/Columns.html), accessed February 2013.
(4) R.W. Slingsby and C.A. Pohl, J. Chromatogr. 458, 241–253 (1988).
(5) P.W. Carr, X. Wang, and D.R. Stoll, Anal. Chem. 81, 5342–5353 (2009).
(6) M.E. Swartz, J. Liq. Chromatogr. Related Technol. 28, 1253-1263 (2005).
(7) C.A. Lucy, J. Chromatogr. A 739, 3–13 (1996).
(8) C.A. Lucy, J. Chromatogr. A 1000, 711–724 (2003).
(9) D.L. Strong, P.K. Dasgupta, K. Friedman, and J.R. Stillian, Anal. Chem. 63, 480–486 (1991).
(10) D.L. Strong, C.U. Joung, and P.K. Dasgupta, J. Chromatogr. 546, 159–173 (1991).
(11) Y. Liu, K. Srinivasan, C. Pohl, and N. Avdalovic, J. Biochem. Biophys. Methods 60, 205–232 (2004).
(12) É. Tyrrell, R.A. Shellie, E.F. Hilder, C.A. Pohl, and P.R. Haddad, J. Chromatogr. A 1216, 8512–8517 (2009).
(13) H. Eghbali, C. Bruggink, Y. Agroskin, C.A. Pohl, and S. Eeltink, J. Sep. Sci. 35, 3461–3468 (2012).
(14) B. Yang, F. Zhang, and X. Liang, Central Eur. J. Chem. 10, 472–479 (2012).
(15) B. Yang, M. Takeuchi, and P.K. Dasgupta, Anal. Chem. 80, 40–47 (2008).
(16) B. Wouters, C. Bruggink, G. Desmet, Y. Agroskin, C.A. Pohl, and S. Eeltink, Anal. Chem. 84, 7212–7217 (2012).
(17) Dionex website (http://www.dionex.com/en-us/products/ion-chromatography/ic-rfic-systems/ics-5000/lp-72594.html?ca=ics5000+), accessed February 2013.
(18) D. Connolly and B. Paull, J. Chromatogr. A 953, 299–303 (2002).
(19) P. Hatsis and C.A. Lucy, Analyst 127, 451–454 (2002).
(20) S. Pelletier and C.A. Lucy, J. Chromatogr. A 1125, 189–194 (2006).
(21) K. Li and H.D.H. Stover, J. Poly. Sci. Part A: Polym. Chem. 31, 3257–3263 (1993).
(22) M.F. Wahab, C.A. Pohl, and C.A. Lucy, J. Chromatogr. A 1270, 139-146 (2012).
(23) C. Liang and C.A. Lucy, J. Chromatogr. A 1217, 8154–8160 (2010).
(24) J.E. Madden, M.J. Shaw, G.W. Dicinoski, N. Avdalovic, and P.R. Haddad, Anal. Chem. 74, 6023–6030 (2002).
(25) B.K. Ng, R.A. Shellie, G.W. Dicinoski, C. Bloomfield, Y. Liu, C.A. Pohl, and P.R. Haddad, J. Chromatogr. A 1218, 5512–5519 (2011).
(26) R.J. Joyce and H.S. Dhillon, J. Chromatogr. A 671, 165–171 (1994).
(27) R. Lin, K. Srinivasan, and C. Pohl, LCGC Europe 24(6), 322–332 (2011).
(28) H.P. Wagner, B.V. Pepich, K.Srinivasan, C. Pohl, B. De Borba, R. Lin, and D.J. Munch, EPA Method 302.0 (2009).
(29) Ayushi, S.D. Kumar, and A.V.R. Reddy, J. Chromatogr. Sci. 50, 81–83 (2012).
(30) P. Zakaria, C. Bloomfield, R.A. Shellie, P.R. Haddad, and G.W. Dicinoski, J. Chromatogr. A 1218, 9080–9085 (2011).
(31) H.P. Wagner, B.V. Pepich, C. Pohl, D. Later, R. Joyce, K. Srinivasan, B. De Borba, D. Thomas, A. Woodruff, and D.J. Munch, EPA Method 314.1 (2005).
(32) S.S. Brudin, R.A. Shellie, P.R. Haddad, and P.J. Schoenmakers, J. Chromatogr. A 1217, 6742–6746 (2010).
Charles A. Lucy is a professor of analytical chemistry at the University of Alberta and is on the Scientific Committee of the International Ion Chromatography Symposium. He is on the editorial advisory boards of Analytica Chimica Acta and Journal of Chromatography A. Direct correspondence to: charles.lucy@ualberta.ca


Charles A. Lucy
M. Farooq Wahab is a senior doctoral student in Dr. Lucy's group at the University of Alberta. His research focuses on developing new stationary phases and high efficiency small particle ion chromatography. Direct correspondence to: mwahab@ualberta.ca

M. Farooq Wahab












. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.




