Adjusting Conditions for a Routine Reversed-Phase HPLC Assay, Part II: Changing Separation Conditions
LCGC North America


When a column is replaced with a new or "equivalent" column, sometimes the chromatogram can change so much that it is no longer suitable for its intended use. In such cases, method adjustment is necessary to correct the change. How much can the chromatographic variables be changed before revalidation is required? What do the regulatory agencies have to say about method adjustment? The authors discuss these issues and propose a technique that can be used to speed selection of new operating conditions.
In the last installment of "Column Watch" (1), the problem of a possible change in column selectivity was introduced, and means for selecting a different column as a replacement were considered. Ten or twenty years ago, changes in separation were not uncommon when replacing a column with one from a different production batch. Personally, we are aware of more than a few such cases within the past decade, in some cases involving columns from major manufacturers. Although batch-to-batch column reproducibility has improved greatly in recent years (2,3), it is premature to claim that all columns currently manufactured will be adequately reproducible for all possible samples and separation conditions. There also is a need to improve some older methods by substituting columns of more recent design, for which the likelihood of encountering major differences in column selectivity is increased greatly.



Ronald E. Majors
Column substitution as described in Part I (1) is likely to work for many cases but not for all. When a change in column does not provide acceptable separation, the only remaining option is to vary separation conditions (mobile-phase composition or temperature). This so-called method adjustment has been shown to be effective generally (4), but it is not used routinely within the pharmaceutical industry because of questions concerning the extent of method revalidation that is required. In this article, we propose a technical review of means for method adjustment and a review of pertinent regulations concerning the required validation of adjusted methods.
Method Development versus Method Adjustment
Method development refers to the initial selection of separation conditions for a proposed assay procedure (5). Method adjustment involves making minor changes in separation conditions for an already developed method, usually as a means of correcting changes in separation selectivity due to a change in column. It should be emphasized that method adjustment is
not
justified for the purpose of correcting errors in mobile phase formulation, sample preparation, or faulty equipment performance. An example of method adjustment is shown in Figure 1. The "original" column yielded the separation of Figure 1a, while Figure 1b shows the separation after replacement of the original column with a new, nominally equivalent column from the same manufacturer. Several changes in relative retention and separation are apparent in these two chromatograms; most notably, the baseline resolution (
R
s
) of peaks 4 and 5 in Figure 1a has degraded to partially overlapping peaks in Figure 1b (arrow). Arbitrary changes in conditions can be made to improve the separation of Figure 1b, as in Figure 1c, where the percentage of acetonitrile in the mobile phase (%B) is increased from 50 to 54.5%. However, relative retention in Figure 1a and 1c is noticeably different. "Complete" method adjustment attempts to achieve a separation that is equivalent to that using the original column, as in Figure 1d, where both temperature and %B have been adjusted. "Equivalent" separation implies generally equivalent band spacing and resolution for each peak; small differences in absolute retention and run time usually can be minimized by further changes in flow rate.

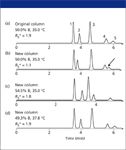
Figure 1: Example of method adjustment. (a) Original column with isocratic conditions as shown, (b) new column, same conditions as in (a), (c) new column, arbitrary adjustment of conditions to restore resolution of (a), and (d) new column, conditions adjusted as described in text. Column: 150 mm 3 4.6 mm C18; mobile phase: acetonitrile-buffer (pH 2.8); flow rate: 2.0 mL/min. Peaks: 1 5 methylbenzoate, 2 5 4-n-pentyl aniline, 3 5 p-nitrotoluene, 4 5 4-n-hexyl aniline, 5 5 4-n-butylbenzoic acid. Adapted from reference 4.
For a change in separation selectivity, only those variables that affect selectivity can be used in method adjustment. Temperature, gradient time, isocratic %B, pH, and buffer or additive concentration are useful variables in this connection. Following the adjustment of separation selectivity, changes in flow rate can be used to minimize differences in run time.
All of the regulatory guidelines stress the importance of system suitability tests as a measure of the ability of an analytical method to produce valid results (for example, reference 6, reference 7, reference 8,). The The United States Pharmacopeia (USP) (reference 8) states that "System suitability tests are an integral part of gas and liquid chromatographic methods. They are used to verify that the resolution and reproducibility of the chromatographic system are adequate for the analysis to be done." As such, it should be obvious that care must be taken to select a system suitability test that indeed discriminates between a method that is operating acceptably and one that is not. The guidelines are not specific about which parameters should be included in system suitability tests. Again, a judgement call is necessary by the method development chemist. In some cases, resolution between two peaks might be the best discriminator, whereas in other cases, minimum peak area or some other criterion also might be appropriate.
An example of selection of system suitability requirements can be taken from the separations of Figure 1. This will help to illustrate the difference between "adequate" separation and "equivalent" separation. System suitability criteria should be based upon the goals of the separation. One such goal for the sample of Figure 1 might be a requirement to be able to adequately quantify each of the five peaks in the sample. One criterion could be that the peaks need to be adequately separated, with the quantitative requirement of Rs > 2.0 for all peak pairs. If this were the system suitability requirement, Figures 1a, 1c, and 1d would pass system suitability and would be expected to give acceptable results for real samples. Another option might be to use relative retention time as the system suitability criterion. For example, the method might require the relative retention of peak 4 to be 0.85-0.95 that of peak 5. In this case, the separations of Figures 1a and 1d pass, but 1c fails. It is clear that the resolution criterion makes the method much more flexible in terms of method adjustment and would be preferred, all other things being equal. We strongly suggest that system suitability tests be designed such that "adequate" separations are acceptable (Figures 1a, 1c, and 1d) rather than requiring "equivalent" separations (Figures 1a and 1d). The system suitability test should discriminate between method performance that will produce acceptable versus unacceptable results, even if the separation does not look exactly the same. Selecting appropriate criteria can be a challenge.
Allowable Changes in Separation Conditions for Method Adjustment
A large change in separation conditions is essentially equivalent to method development, as opposed to "smaller" changes in method adjustment. Provisional recommendations for maximum changes in method adjustment have been proposed, assummarized in Table I.

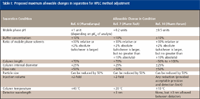
Table I: Proposed maximum allowable changes in separation for HPLC method adjustment
United States Food and Drug Administration (FDA) guidelines for bioanalytical (analysis of drugs in biological matrices) method validation (6) do not directly address the requirements for revalidation following method adjustment. The guidelines imply that as long as system suitability passes for the method, it is acceptable to use. It is our opinion that modifications to method conditions that fall within the parameters of Table I do not require revalidation. More extensive changes, however, would likely fall under the category of "partial validation" (6), for which one batch is run to confirm that the required levels of precision and accuracy are maintained under the new conditions.
The FDA guidelines for analytical procedures for drug substance (pure drug) and drug product (formulated drug) (12) are more specific than the bioanalytical guidelines (6). This guidance states that revalidation requirements depend upon the nature of the change. Again, system suitability is used as the criterion: "If during each use an analytical procedure can meet the established system suitability requirements only with repeated adjustments to the operating conditions . . . the analytical procedure should be . . . revalidated" (12). Reference is made to use of compendial procedures (for example, USP), so it is our opinion that the adjustments of Table I would not require revalidation, provided system suitability passes the acceptance criteria.
International Committee on Harmonization (ICH) guidelines (7,13) are similar to FDA guidelines (12). These state that the revalidation requirements depend upon the nature of the change (13) and on system suitability as the performance criterion (7); reference is made to pharmacopoeias for additional information (7). As such, these guidelines also imply that if the adjustments fall within the criteria of Table I, no revalidation is required.
The USP guidelines (8) historically have relied heavily upon system suitability tests as a means of determining adequate method performance. They state, "Adjustments of operating conditions to meet system suitability requirements may be necessary," (8) but do not comment on revalidation requirements. More specific guidelines appear likely in the near future. Pharmacopeial Forum is used to propose changes to the USP. A recent Forum article (11) proposed the acceptance limits for parameter adjustment shown in Table I. These generally are similar to the preceding recommendations (9,10), which it is likely were used as the basis for the Forum recommendations.
From the aforementioned review, three consistent threads are apparent. First, system suitability should be the primary criterion for the acceptability of method performance. Any change that is made such that system suitability does not pass should be a red flag for the operator. In such cases, the method should be further modified until system suitability again passes. Second, the adjustment limits of Table I (especially those of reference 11 when incorporated into the USP) generally should be acceptable for method adjustment, requiring no revalidation provided system suitability passes. Finally, the judgement of the user is very important in the decision of whether or not to revalidate the method. The guidelines of Table I, internal company policy (standard operating procedures) and common sense should guide users in the adjustment versus revalidate decision. In any event, proper documentation should be made for any method changes.
Validation Versus Revalidation
In the context used in the present discussion, we consider validation to refer to the initial proof that the method meets the performance requirements set by users and regulatory agencies. Validation might include experiments that are not directly related to the chromatography, such as stability of stock solutions, sample stability, or extraction recovery. We refer to revalidation as the process that shows a modified method still meets the initial performance requirements. As such, repeat of nonchromatographic experiments, such as solution stability, have no benefit and are not performed. Similarly, the number of runs required for statistical support of method performance during validation might not be necessary for revalidation.
Regulatory guidelines are not clear regarding the level of revalidation that is required, with the exception of the bioanalytical guidelines (6), which require, as a minimum, a single run to show that acceptable method performance can be obtained. As a result of the vagueness of the guidelines, most of the justification for revalidation (or not) lies with the user. For any revalidation of a method subject to regulatory guidelines, a written revalidation plan should be prepared, including predetermined acceptance criteria. It is expected that common sense will be used in conjunction with the guidelines of Table I when designing the revalidation experiments. For example, a change in mobile phase organic solvent or pH that exceeds the suggestions of Table I is more likely to have a negative impact on the method than a change in flow rate or column diameter.
Means for Efficient Method Adjustment
Method adjustment can be carried out in similar fashion as for method development (5), but with the proviso that only "small" changes in conditions are allowed. A systematic procedure for method adjustment has been described (4), based upon the observation that changes in separation as a result of change in two or more conditions are additive, when the change in each condition is restricted within certain limits (14). This means that given separations for the original and new columns, only one additional experiment is needed for each condition (and either column) to optimize method adjustment. For example, if temperature and %B were the variables under consideration, data for a run at one temperature and one %B on both the original and the new column would be combined with a third run at a second temperature (with the original %B) and a fourth run with a new %B (and the original temperature) on either column. These data would be used to determine the best combination of temperature and %B to use on the new column for equivalent separation. Commercial software for carrying out method adjustment as described in (4) is available (Method Match, Rheodyne LLC, Rohnert Park, California), with the result of Figure 1d. Note that the required changes in %B (-0.7% or -1.4% relative) and temperature (+2.8 °C) for this example are well within the limits of Table I.
The effectiveness of method adjustment as described in (4) was evaluated by comparing the isocratic separation of approximately 60 different samples on two nominally equivalent columns, which differed in carbon percentage by 10% (and gave significantly different separation). These samples contained 5-15 components each, and method adjustment was carried out by varying temperature and %B only. The separation of each sample on the two columns was compared in terms of the coefficient of variation (CV) of resolution Rs, with a value of CV ⤠15% being regarded as "equivalent" (for example, a change in Rs from 2.0 to 1.7). Ten different samples with the same number of components were grouped, and an average CV for the group determined. Data points above the line for CV = 15% in Figure 2 correspond to possibly unacceptable separation; it is seen that all of the unadjusted separations (solid squares) fail this criterion. Average values of CV are plotted in Figure 2 versus the number of components in the sample. As expected, the probability of an accep adjustment decreases as the number of sample components increases, with a sample containing approximately 14 components having an even chance of a successful adjustment. These results suggest that method adjustment based on change in temperature and %B generally will be successful, especially for samples containing no more than 14 components. The probability of a successful adjustment increases if additional separation conditions are varied (for example, pH or buffer concentration).

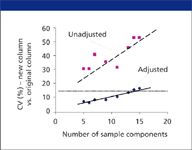
Figure 2: Efficacy of method adjustment for approximately 60 randomly selected samples, changing only temperature and %B. CV values for differences in resolution between original and replacement. Data points for each value of n are an average of 6-10 samples each. Adapted from reference 4.
Conclusions
The challenge of obtaining adequate method performance across several years can be complicated by unexpected changes in column chemistry for nominally equivalent columns (same part number) over time. This can be further complicated when a column is discontinued or replaced with an "improved" version. In Part I (1) of this series, we described a technique to select columns likely to give equivalent separations. In the current discussion, we have addressed the issue of methods that are observed to change slightly when a replacement column (either of the original type or an "equivalent" column) is installed. The regulatory guidelines depend heavily upon system suitability tests as a discriminator of method performance. In addition, there exist limits (Table I) within which variation in chromatographic parameters are allowed by regulatory agencies (assuming the recommendations of reference 11 are adopted by the
USP
). When method changes are made that fall within the parameters of Table I, we consider this as method adjustment, and little if any revalidation is required, so long as system suitability passes. Method changes that exceed the guidelines of Table I are more likely to be considered modifications of the method that will require some degree of revalidation.
References
(1) L.R. Snyder and J.W. Dolan,
LCGC
22
(12), 1146-1152 (2004).
(2) U.D. Neue, E. Serowik, P. Iraneta, B.A. Alden, and T.H. Walter, J. Chromatogr., A 849, 87 (1999).
(3) M. Kele and G. Guiochon, J. Chromatogr., A 869, 181 (2000).
(4) J.W. Dolan, L.R. Snyder, T.H. Jupille, and N.S. Wilson, J. Chromatogr., A 960, 51 (2002).
(5) L.R. Snyder, J.J. Kirkland, and J.L. Glajch, Practical HPLC Method Development, 2nd ed. (WileyInterscience, New York, 1997).
(6) "Guidance for Industry: Bioanalytical Method Validation," FDA (May 2001).
(7) "Guidance for Industry: Q2B Validation of Analytical Procedures: Methodology," ICH (November 1996).
(8) The United States Pharmacopeia, USP 28, Rockville, Maryland (2005).
(9) R. Cox and G. Menon, PharmEuropa 10, 58 (1998).
(10) W.B. Furman, J.G. Dorsey, and L.R. Snyder, Pharm. Technol., 58 (June 1998).
(11) Pharmacopeial Forum 30(3), 1015-1017 (May/June 2004).
(12) "Guidance for Industry: Analytical Procedures and Methods Validation," FDA (August 2000).
(13) "Guideline for Industry: Text on Validation of Analytical Procedures," ICH-Q2A (March 1995).
(14) J.W. Dolan, D.C. Lommen, and L.R. Snyder, J. Chromatogr. 535, 55 (1990).
Lloyd R. Snyder is with LC Resources, 26 Silverwood Ct., Orinda, CA 94563. He can be reached at snyder0036@comcast.net.
John W. Dolan is with BASi Northwest Laboratory, McMinnville, OR 97128. He can be reached at John.Dolan@Bioanalytical.com
Ronald E. Majors"Column Watch" Editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, Delaware, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.


















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



