Why Use Signal-To-Noise As a Measure of MS Performance When It Is Often Meaningless?
Special Issues
Signal-to-noise of a chromatographic peak from a single measurement has been used determine the performance of two different MS systems, but this parameter can no longer be universally applied and often fails to provide meaningful estimates of the instrument detection limits (IDL).
In the past, the signal-to-noise ratio of a chromatographic peak determined from a single measurement served as a convenient figure of merit used to compare the performance of two different mass spectrometry (MS) systems. The evolution in the design of MS instrumentation has resulted in very low noise systems that have made the comparison of performance based upon signal-to-noise increasingly difficult, and in some modes of operation, impossible. This is especially true when using ultralow-noise modes, such as high-resolution MS or tandem MS, where there often are no ions in the background and the noise is essentially zero. This occurs when analyzing clean standards used to establish the instrument specifications. Statistical methodology that is commonly used to establish method detection limits for trace analysis in complex matrices is a means of characterizing instrument performance that is rigorously valid for both high and low background noise conditions. Instrument manufacturers should begin to provide customers an alternative performance metric, in the form of instrument detection limits based on the relative standard deviation of replicate injections, to allow analysts a practical means of evaluating an MS system.
For decades, signal-to-noise ratio (S/N) has been a primary standard for comparing the performance of chromatography systems including gas chromatography–mass spectrometry (GC–MS) and liquid chromatography (LC)–MS. Specific methods of calculating S/N have been codified in the U. S., European, and Japanese Pharmacopeia (1–3) to produce a uniform means of estimating instrument detection limits (IDL) and method detection limits (MDL). The use of S/N as a measure of IDL and MDL has been useful, and is still in use, for optical-based detectors for LC, and flame-based detectors for GC. As MS instrument design has evolved, the ability to accurately compare performance based on S/N has become increasingly difficult. This is especially true for trace analysis by MS using ultralow-noise modes, such as high-resolution mass spectrometry (HRMS) or tandem MS (MS-MS). S/N is still a useful parameter, particularly for full scan (EI) MS, but the comparison of high-performance MS analyzers should be based upon a metric that is applicable to all types of MS instruments and all operating modes. Statistical means have long been used to establish IDL and MDL (4–7) and are suitable to all operating modes for mass spectrometers.
Evolution of Instrumentation
Many sources of noise have been reduced by changes to the MS design such as low noise electronics, faster electronics allowing longer ion sampling (signal averaging), modified ion paths to reduce metastable helium (neutral noise), and signal processing (digital filtering). HRMS and MS-MS are also effective means to reduce chemical background noise, particularly in a complex sample matrix. For the signal, a longer list of improvements to the source, analyzer, and detector components have resulted in more ions produced per amount of sample. In combination, the improvements in S/N and the increase in sensitivity have resulted in significant and real lowering of the IDL and MDL.
Lack of Guidelines for S/N Measurements
The measurement of the "signal" is generally accepted to be the height of the maximum of the chromatographic signal above the baseline (Figure 1). However, some of the changes in GC–MS S/N specifications have been artificial. The industry standard for GC–electron ionization (EI) MS has changed from methyl stearate, which fragments extensively to many lower intensity ions, to compounds that generate fewer, more intense ions, such as hexachlorobenzene (HCB) and octafluoronaphthalene (OFN). The change to OFN held a secondary benefit for noise: the m/z 272 molecular ion is less susceptible to baseline noise from column bleed ions (an isotope of the monoisotopic peak of polysiloxane at m/z 281 increased baseline noise for HCB at m/z 282).


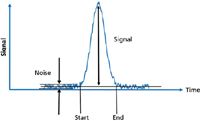
Figure 1: Analyte signal as a function of time for a chromatographic peak, demonstrating the time dependence in the amount of analyte present.
Improvements in instrument design and changes to test compounds were accompanied by a number of different approaches to measuring the noise. In the days of strip chart recorders and rulers, a common standard for noise was to measure the peak-to-peak (minimum to maximum) value of baseline noise, away from the peak tails, for 60 s before the peak (Figure 1) or 30 s before and after the peak. As integrator and data systems replaced rulers, the baseline segments for estimation of noise were autoselected and noise was calculated as the standard deviation (STD) or root-mean-square (RMS) of the baseline over the selected time window.
Automation of the calculation holds many conveniences for a busy laboratory, but the noise measurement criteria have become less controlled. In some cases, vendors have estimated noise based on a very narrow window (as short as 5 s) and location that can be many peak widths away from the peak used to calculate signal. These variable "hand-picked" noise windows make it easy for a vendor to claim a higher S/N by judiciously selecting the region of baseline where the noise is lowest. Generally, the location in the baseline where the noise is calculated is now automatically selected to be where the noise is a minimum. Figure 2 shows three different values of RMS noise calculated at different regions of the baseline. The value of the noise at positions a, b, and c is 54, 6, and 120, and the factor of 20 variation in S/N is exclusively because of where in the baseline the noise is measured. However, these S/N values will not correlate to the practical IDL for automated analyses that the laboratory performs. Therefore, the use of signal-to-noise as an estimate of the detection limit will clearly fail to produce usable values when there is low and highly variable ion noise and the choice of where to measure it is subjective.

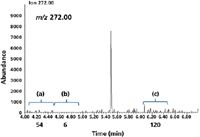
Figure 2: EI full-scan extracted ion chromatograms of m/z = 272 from 1 pg OFN.
The situation becomes even more indeterminate when the background noise is zero, as shown in the MS-MS chromatogram in Figure 3. In this case, the noise is zero and S/N then becomes infinite. The only "noise" observed in Figure 3 is because of the electronic noise, which is several orders of magnitude lower than noise because of the presence of ions in the background. This situation can be made more severe by increasing the threshold for ion detection. Under these circumstances, it is possible to increase the ion detector gain, and hence the signal level, without increasing the background noise. The signal of the analyte increases, but the noise does not. This is obviously misleading since the signal increased, but there was no increase in the number of ions detected, and therefore, no change to the real detection limit. This allows S/N to be adjusted to any arbitrary value without changing the real detection limit.


Figure 3: EI MS-MS extracted ion chromatogram of m/z 222.00 from 1 pg OFN exhibiting no chemical ion noise.
Two decades ago, instrument specifications often described the detailed analytical conditions that affected the signal or noise value, such as chromatographic peak width, data rate, and time constant. Today, those parameters are missing or hard to find, even though a novice chromatographer realizes a narrow peak will have a larger peak height than a broad peak. In many cases, the GC conditions are selected to make the chromatographic peak extremely narrow and high, with the result that the peak is under-sampled (only one or two data points across a chromatographic peak). This may increase the calculated S/N, but the under-sampling degrades the precision and would not be acceptable for most quantitative methods. Once again, a false perception of performance has been provided. Accurate, meaningful comparison of instrument performance for practical, analytical use based on published specifications is increasingly difficult, if not impossible. Alternative means are required to determine the instrument performance and detection limit, which is generally applicable to all modes of MS operation.
What Is the Alternative for S/N?
S/N is still useful and represents a good first estimate to guide other statistically based estimates of performance. Every analytical laboratory should understand and routinely use statistics to evaluate and validate their results. The analytical literature has numerous articles that utilize a more statistical approach for estimating an IDL or MDL. The U.S. EPA has dictated a statistical approach to MDL, and can be found in the recommended EPA "Guidelines Establishing Test Procedures for the Analysis of Pollutants" (6). The European Union also has supported this position. A commonly used standard in Europe is found in "The Official Journal of the European Communities;" Commission Decision of 12 August 2002; Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (8).
Both of these methods are generally similar and require injecting multiple replicate standards to assess the uncertainty in the measuring system. A small number of identical samples having a concentration near the expected limit of detection (within 5–10) are measured, along with a comparable number of blanks. Because of the specificity of MS detection, the contribution from the blank is negligible and is often excluded after the significance of the contribution has been confirmed. The mean value X mean and standard deviation (STD) of the set of measured analyte signals, Xn (that is, integrated areas of the baseline subtracted chromatographic peaks), is then determined. Even for a sensitivity test sample, the "sampling" process is actually a complex series of steps that involves drawing an aliquot of analyte solution into a syringe, injecting into a GC system, and detecting by MS. Each step in the sampling process can introduce variations in the final measured value of the chromatographic peak area, resulting in a sample-to-sample variation or "sampling noise." These variances will generally limit the practical IDL and MDL that can be achieved.
The variance in the measured peak areas includes the analyte signal variations, the background noise, and the variance from injection to injection. Distinguishing the statistical significance of the mean value of the set of measured analyte signals from the combined system and sampling noise can then be established with a known confidence level. The IDL is then the smallest signal or amount of analyte that is statistically greater than zero within a specified probability of being correct. The IDL (or MDL) is related (9,10) to the standard deviation STD of the measured area responses of the replicate injections and a statistical confidence factor tα by
IDL = (tα) (STD), where the STD and IDL are expressed in area counts.
Alternately, many data systems report relative standard deviation (RSD = STD/Mean Value). In this case, the IDL can be determined in units of the amount of the standard (ng, pg, or fg) injected by
IDL = (tα) (RSD) (amount standard)/100%

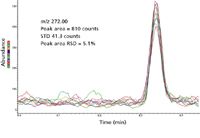
Figure 4: EI full-scan extracted ion chromatograms of m/z = 272 from 200 fg OFN, eight replicate injections, 3.3 Hz data rate.
When the number of measurements n is small (n < 30), the one-sided Student t-distribution (11) is used to determine the confidence factor tα. The value of tα comes from a table of the Student t-distribution using n–1 (number of measurements minus one) as the degrees of freedom, and 1–α is the probability that the measurement is greater than zero. The larger the number of measurements n, the smaller is the value of tα, and less the uncertainty in the estimate of the IDL or MDL. Unlike the statistical method of determining IDL and MDL, the use of signal-to-noise from a single sample measurement does not capture the "sampling noise" that causes multiple measurements of the same analyte to be somewhat different.
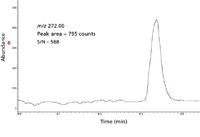
Figure 5: EI full-scan extracted ion chromatograms of m/z = 272 from 200 fg OFN, first entry in Table I, 3.3 Hz data rate.
As an example, for the eight replicate injections in Figure 4 (n = 7 degrees of freedom) and a 99% (1–α = 0.99) confidence level, the value of the test statistic from the t-table is tα = 2.998. For the eight 200-fg samples, the mean value of the area is 810 counts, the standard deviation is 41.31 counts, and the value of the IDL is: IDL = (2.998)(41.31) = 123.85 counts. Since the calibration standard was 200 fg and had a measured mean value of 810 counts, the IDL in femtograms is: (123.85 counts)(200 fg)/(810 counts) = 30.6 fg. Alternately in terms of RSD, the IDL = (2.998)(5.1%)(200 fg)/100 = 30.6 fg. Thus, an amount of analyte greater or equal to 30.6 fg is detectable and distinguishable from the background with a 99% probability. In contrast, S/N measured on one single chromatogram (Figure 5) would give an IDL of 1.1 fg, assuming that IDL = 3 × rms noise (first entry in Table I). The high S/N is the result of a commonly used algorithm that calculates the noise using the lowest noise sections of the baseline adjacent to the peak where the noise is unusually low. The individual S/N and IDL values measured for each injection in Figure 4 are tabulated in Table I.

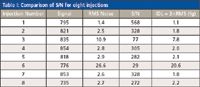
Table I: Comparison of S/N for eight injectionsThe IDL values range from 1.1 fg to 20.5 fg, and even the largest value is significantly smaller than the IDL determined by statistical means, which is more realistic.
Estimating Relative Sensitivity
A secondary benefit to specifying detection limits in statistical terms is to use the RSD as an indirect measure of the relative number of ions on a chromatographic peak. It is known (12) that if a constant flux of ions impinge on a detector and the mean number of ions detected in a particular time interval averaged over many replicate measurements is N, then the RSD in the number ions detected is
1/√N
Hence, decreasing the number of ions in a chromatographic peak will increase the area RSD. This effect can be seen by comparing the RSD in Figure 6 and Figure 4 where the analyte amounts are 1 pg and 200 fg, respectively. The increased RSD at the lower sample amount is, in large part, because of ion statistics. Five times lower number of ions means
√5=2.24
times increase in RSD. It follows that the 2.1% RSD at 1 pg will become 4.7% at 200 fg just from ion statistics. It is not unexpected that, at lower sample amounts, there also will be some additional variance from the chromatography. Thus, RSD measurements of the same amount of analyte on two different instruments can be used to indicate the relative difference in sensitivity near the detection limit, assuming that other contributions to the total variance of the signals are small. The more sensitive instrument will have the smaller RSD if all other factors are the same (that is, peak width and data rate). This avoids the uncertainty of inferring sensitivity from the measurement of peak areas where there is no baseline noise.

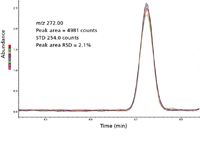
Figure 6: EI full-scan extracted ion chromatograms of m/z = 272 from 1 pg OFN, eight replicate injections, 3.3 Hz data rate.
Impact of Changing from S/N to RSD
For MS vendors, there is complexity and cost in providing customers this valuable information. If the GC is configured with an autosampler and a splitless inlet, it should be fairly simple to add 7–10 replicate injections. This might add a little time to the installation, but the use of an autosampler should keep this at a minimum. But how do you test a system that does not have an autosampler or the appropriate inlet? Manual injections will add operator-dependent imprecision to measuring the peak area. Reproducible injection using a manually operated injector device like the Merlin MicroShot Injector (Merlin Instrument Co., Half Moon Bay, California), are necessary to reduce the sampling noise. If the method uses headspace or gas analysis, it could be costly to test as a liquid injection and then re-configure for the final analysis. These costs must be managed appropriately, but factors like cost and complexity for some configurations should not prevent a transition to a better, statistically based standard such as %RSD for the majority of the systems that do not have these inlet limitations.
From the customer's perspective, however, there is a significant benefit to having a system level test of performance that offers a realistic estimate of the IDL and the system precision near the limits of detection where it is the most critical. The purchase of a mass spectrometer is a significant capital expenditure, and often purchasing decisions are based on a customer's specific application need. Instrument manufacturers do not have the resources to demonstrate their product's performance for every application. It would be valuable to have a simple, but realistic, means of evaluating a particular MS instrument's performance in the form of the IDL and the system precision. These will form lower bounds to the MDL and method precision for specific applications.
Summary
In the past, the S/N of a chromatographic peak determined from a single measurement has served as a convenient figure of merit used to compare the performance of two different MS systems. However, as we have seen, this parameter can no longer be universally applied and often fails to provide meaningful estimates of the IDL. A more practical means of comparing instrument performance is to use the multi-injection statistical methodology that is commonly used to establish MDLs for trace analysis in complex matrices. Using the mean value and RSD of replicate injections provides a way to estimate the statistical significance of differences between low level analyte responses and the combined uncertainties of the analyte and background measurement, and the uncertainties in the analyte sampling process. This is especially true for modern mass spectrometers for which the background noise is often nearly zero. The RSD method of characterizing instrument performance is rigorously and statistically valid for both high and low background noise conditions. Instrument manufacturers should start to provide customers with an alternative performance metric in the form of RSD-based instrument detection limits to allow them a practical means of evaluating an MS system for their intended application.
References
(1) European Pharmacopoeia 1, 7th Edition.
(2) United States Pharmacopeia, XX Revision (USP Rockville, Maryland, 1988).
(3) Japanese Pharmacopoeia, 14th Edition.
(4) ASTM: Section E 682–93, Annual Book of ASTM Standards, 14(1).
(5) P.W. Lee, Ed. Handbook of Residue Analytical Methods for Agrochemicals 1, Chapter 4 (2003).
(6) U.S. EPA – Title 40: Protection of Environment; Part 136 – Guidelines Establishing Test Procedures for the Analysis of Pollutants; Appendix B to Part 136 – Definition and Procedure for the Determination of the Method Detection Limit – Revision 1.11.
(7) "Uncertainty Estimation and Figures of Merit for Multivariate Calibration," IUPAC Technical Report, Pure Appl. Chem. 78(3), 633–661 (2006).
(8) Official Journal of the European Communities; Commission Decision of 12 August 2002; Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results.
(9) "Guidelines for Data Acquisition and Data Quality Evaluation in Environmental Chemistry," ACS Committee on Environmental Improvement Anal. Chem. 52, 2242–2249 (1980).
(10) "Signal, Noise, and Detection Limits in Mass Spectrometry," Agilent Technologies Technical Note, publication 5990–7651EN.
(11) D.R. Anderson, D.J. Sweeney, and T.A. Williams, Statistics (West Publishing, New York, 1996).
(12) P.R. Bevington and D.K. Robinson, Data Reduction and Error Analysis for the Physical Sciences (WCB McGraw-Hill, Boston, 2nd Edition, 1992).
Greg Wells, Harry Prest, and Charles William Russ IV are with Agilent Technologies, Inc., Santa Clara, California.
















University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.


Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

