What’s Trending in LC Troubleshooting?


We look at problems in LC that can be traced to contaminated solvents or reagents, and how sample solvent effects can lead to poor performance in HILIC
The challenges we face in troubleshooting problems with LC separations are highly diverse. This month we take a closer look at topics that have garnered more attention recently.
As I look through the topics that have been discussed in this column over the past 30+ years, and think about our day-to-day experiences with modern liquid chromatography (LC), it is clear that there are some troubleshooting topics that just continue to hang around. Although pump check valves have improved over the years, they still fail occasionally, and are often found to be the cause of unstable flow from LC pumps. Vast improvements have been made in LC column technologies over the years, but still it is not uncommon to observe symptoms of failed columns, including split or tailed peaks, or both. For this month's column, I decided to pay close attention to what is trending in LC troubleshooting at the moment. I spent some time browsing online LC discussion boards, where people are describing their problems and others are offering advice. At conferences I have attended recently, I listened more closely to the challenges people described in their conference talks and during the coffee breaks. First, I have to say that an incredible diversity of problems is addressed in the online discussion groups. LC is an amazingly powerful analytical tool, but, looking at a discussion board, we realize just how many ways it can break, and that we are still quite a ways off from having truly autonomous LC systems. Based on what I've seen and heard recently, this month I decided to focus on two particular topics: problems in LC that can be traced to contaminated solvents or reagents, or both; and the effect of sample solvent that can lead to poor performance of HILIC separations.
The Problem Cannot Be Caused by the Solvent, Can It?
We are fortunate to be able to buy high quality solvents for use in chromatography and mass spectrometry, typically at remarkably low cost. We are able to purchase concentrated acids with extremely low levels of metals for doing trace-level metal analysis, and solvents like acetonitrile, water, and methanol for liquid chromatography–mass spectrometry (LC–MS) that have very low levels of small organic contaminants. The quality of these materials obtained from reputable manufacturers tends to be highly consistent. However, it does not always work out this way. Sometimes these reagents are contaminated during manufacture, storage, or at the point of use, so we should be careful to consider in the course of troubleshooting LC and LC–MS systems that a contaminated solvent or other reagent could be the cause of a major problem. I think sometimes the good experiences we have with high quality reagents most of the time can lull us into a false sense of security, such that, in the course of normal troubleshooting, potential problems with solvents and reagents are not among the first things to enter our minds.
Examples of Experiences with Contaminated Solvents and Reagents
Given enough time in a laboratory, eventually everyone will encounter a situation in which a contaminated reagent results in a significant problem with a method. All laboratories with substantial experience have stories to tell about such problems. In the examples below, I share some of the problems I've encountered in my own work, as a way of encouraging you to seriously consider the possibility that future problems could be due to a contaminated solvent or reagent.
My most recent encounter with a contaminated solvent occurred in the context of an LC–MS experiment where we were using premixed 0.1% formic acid in water in 4-liter amber bottles from a reputable distributor. We observed ions in the mass spectrometer typically associated with polyethylene glycol (PEG), and the abundance of these ions decreased as we carried out a solvent gradient elution program in reversed-phase mode (that is, the PEG ions decreased as the fraction of the aqueous solvent in the mobile phase decreased over time). The PEG peaks were so intense that they dominated the total ion current, making it difficult to detect any peaks from the peptide mixture we had injected. Upon switching to a different bottle of 0.1% formic acid in water (from the same vendor, but with a different lot number), we immediately saw the signals for the PEG ions drop to the point where they were hardly detectable after 30 min of flushing the contaminated solvent from the system.
We have also observed problems with neat formic acid as a mobile-phase additive. I wrote about this previously in the context of different sources of contaminants in mass spectrometry experiments specifically (1), but I think it is worth repeating here. A few years ago we faced a situation in our work with LC–MS analyses of intact proteins where we had great results on a Friday, and no MS signal for the proteins at all the following Monday. After pursuing many other possible causes for this dramatic change, we found that the cause of the loss in signal was a "bad" bottle of neat formic acid that we were using as additive for the aqueous component of our mobile phase. To this day, we have no idea what that formic acid was contaminated with that killed our MS signal, but changing to a different source of formic acid immediately restored the performance of the method to normal. We never suspected that the reagent could be the cause of the problem in this case because the bottle was brand new, and was marketed as an "additive for LC–MS."
Several years ago, I did some work with an instrument manufacturer investigating a change observed in the magnitude of change in the baseline of an ultraviolet-visible (UV-vis) absorbance detector upon changing solvents. Initially, we had the idea that the baseline changes were somehow related to differences in the refractive indices of the solvents used, and that the change in baseline was somehow related to the optical properties of some of the detector components. In the end, however, we found that the cause of the observed changes was related to significant differences between the levels of UV-absorbing solvent contaminants in different manufacturing lots of solvent. In this case, the problem was not so much that the solvent was not within specification for UV absorbance at low wavelength, but that there was so much variability from one lot to the next.
Strategies to Support Troubleshooting Potential Problems with Solvents and Reagents
In the course of validating a process (for example, a chromatographic method) in a regulated environment, it is typical to evaluate multiple lots of critical materials (for instance, chromatographic columns) to avoid the negative consequences of unanticipated variation in the process. In less regulated environments, it is tempting to skip over these details in the interest of saving time and resources. However, having learned the hard way how frustrating and time consuming reagent-related challenges can be, in my laboratory we have become more diligent about safeguarding against these problems. These steps will certainly look different in other laboratories, but for us the important guidelines are as follows:
1. Retain portions of "old" reagents that are known to work well when switching to a new vendor or part number. This gives us the ability to double-check the functionality of the rest of the system in an event where we suspect a problem with a new reagent.
2. Establish solid benchmarking data for critical reagents. I've written about this in the past concerning monitoring LC column performance (2), but the idea applies here as well. Collect baseline data that reflect characteristics of the reagent that are meaningful to the method. For example, we characterize formic acid that we use as a mobile- phase additive both in terms of contaminants that are detectable by MS under typical LC–MS conditions, and in terms of the effect of the additive on detection sensitivity for proteins and peptides. These baseline data, updated periodically over time, gives us a reliable way to assess the effect of switching to a new source of the reagent.
Mind the Sample Solvent!
A problem that I have been hearing and reading about a lot recently, in a variety of places, is the observation of "no retention" under HILIC conditions. This most definitely can be a real problem where the analyte of interest is simply too soluble in the mobile phase, relative to the stationary phase, and no useful amount of retention is observed. However, I think in many cases the observation of no retention has more to do with the effect of the sample solvent (or sample diluent) on the behavior of the analyte in the column than it does with the inherent retention of that analyte in the mobile phase. In these cases, simply adjusting the sample solvent or injection volume, or both, can have a significant effect on the chromatography, turning an awful separation into a good one.
Although I think the impact of the sample solvent on LC separation performance is generally underappreciated, it has at least been studied extensively in the context of reversed-phase separations (3–5). And because most LC practitioners do more reversed-phase separations than anything else, it is perhaps most easily understood in this context as well. Figure 1 shows a comparison of the chromatograms obtained for a sample of simple alkylphenones injected in solvents containing more (b)or less (a) acetonitrile than the mobile phase, but under conditions that are otherwise identical. Clearly the effect of the sample solvent is dramatic. I think the simplest way to understand what is going on here is to recognize that, in this case, the injection volume of 40 µL is about 70% of the dead volume of the column itself. This means that when all of the sample has entered the column from the injector, literally 20 mm of the 30 mm column length is filled with sample. In other words, for this 20 mm segment of the column, the sample solvent is the mobile phase. Under these conditions, the retention factor of even the most retained compound in the mixture is just 3, meaning that most of the compounds simply travel with the band of sample solvent all of the way from the column inlet to the column outlet. There is no chance for any useful separation to occur because the analytes are not retained enough to be first separated from the band of sample solvent, and then be separated from each other.


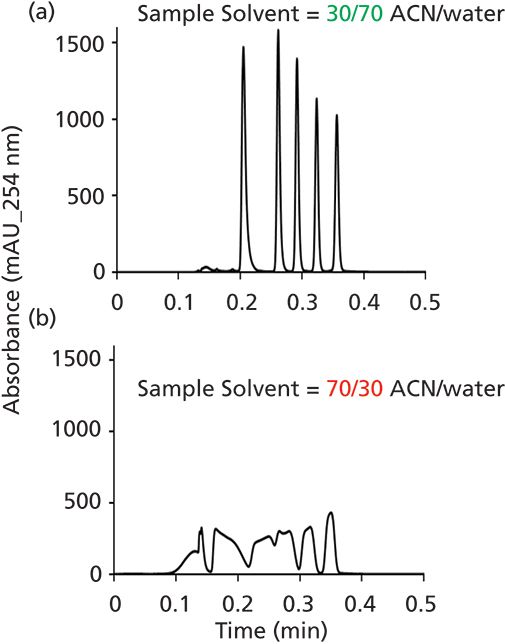
Figure 1: Comparison of chromatograms obtained from injection of samples in a) 30:70 (v/v) acetonitrile:water, or b) 70:30 (v/v) acetonitrile:water. Conditions: column, 30 mm × 2.1 mm i.d. C18; injection volume, 40 µL; gradient elution from 50 to 90% acetonitrile was from 0 to 15 s, with water as the aqueous phase; flow rate, 2.5 mL/min. Analytes are alkylphenone homologs from acetophenone to hexanophenone.
The same kind of thing can happen in HILIC separations, except that in HILIC the discussion should be focused more on the water content of the sample than on the organic solvent content. The effects of injection volume and the water content and organic solvent type in the sample have been described in the literature, although not in the same depth as for reversed-phase separations (6–9). Figure 2 shows a series of chromatograms obtained under HILIC conditions using a bare silica phase for the separation of cytidine and guanosine. When the injection volume is small (top row of Figure 2), the sample composition has little effect on the resolution of this peak pair, and when the sample contains a large fraction of acetonitrile (left column of Figure 2), the injection volume has little effect on resolution. However, when the injection volume is large (bottom row of Figure 2) the sample composition is very important; likewise, when the sample contains a significant amount of water (right column of Figure 2), the injection volume is important. The worst-case scenario involving a large injection of a sample containing a large fraction of water leads to a disastrous result (lower right corner of Figure 2). Indeed, it would appear under this condition that there can be no retention of cytidine and guanosine at all (see peak at dead time indicated by the blue arrow). But, in fact, this is a kind of artifact caused not by low retention in the mobile phase, but by an inappropriate combination of injection volume and sample solvent composition.

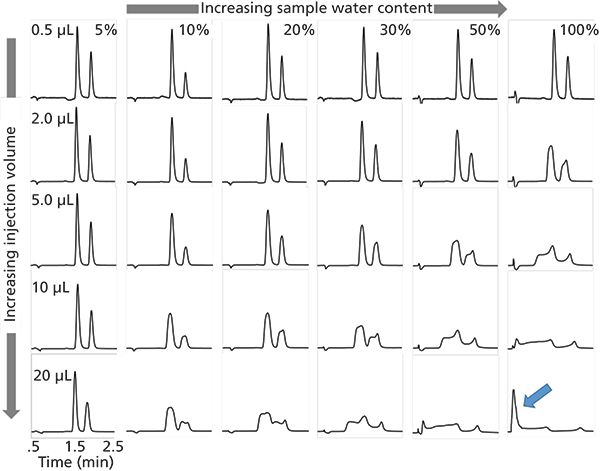
Figure 2: Effect of injection volume and sample solvent composition on peak shape and resolution for hydrophilic interaction chromatography (HILIC) separations of highly water soluble compounds. Chromatographic conditions: column, 100 mm × 2.1 mm i.d. Ascentis Express HILIC (2.7 µm); flow rate, 0.4 mL/min.; temperature, 40 °C; mobile phase, 90:10 acetonitrile-100 mM ammonium formate adjusted to 3.0 with formic acid; detection, ultraviolet (UV) absorbance at 254 nm. Samples were prepared in ACN and water, and contained 50 and 40 µg/mL cytidine and guanosine, respectively. Signals are normalized for each injection such that each injection volume produces a peak of the same height if the peak shapes are the same.
Strategies for Minimizing Sample Solvent Effects in HILIC Separations
If we understand that the combination of injection volume and sample solvent can have a significant negative impact on separations, then the question becomes, How do we avoid this problem? The simplest possible solution is to just inject smaller volumes of samples. Generally speaking if the injection is less than 1% of the column dead volume, the sample solvent will not have much of an effect on the separation. As always, there will be exceptions to this and you may have to go down to 0.1%, but I think 1% is a good rule of thumb. However, simply reducing the injection volume cannot be a viable solution in all situations, because injecting such small amounts will frequently lead to inadequate detection sensitivity (that is, poor detection limits). In other words, in applications where we need to be able to detect low concentrations of analyte, we need to find ways to inject larger volumes of sample without negatively affecting the separation itself. One approach is to dilute samples that are high in water content with organic solvent, preferably acetonitrile. This is analogous to what is commonly done for reversed-phase separations of samples high in organic solvent content (4,5), and can be very effective. However, I think the biggest problem with this approach is that some polar analytes of interest to HILIC separations are not very soluble in acetonitrile-rich solvents. A good example of a situation like this involves separations of proteins by HILIC. We have done quite a bit of this type of work in my laboratory, and we find that the mobile phases that work for these separations require about 65-70% ACN. But, if we add enough ACN to our aqueous sample to get to 70% acetonitrile, the protein crashes out of solution pretty quickly. There are at least two known approaches to handle this type of sample. The first involves starting the solvent elution program used for the HILIC separation at a much higher percentage of acetonitrile than is needed for the separation itself (10). When the highly aqueous sample is injected into this mobile phase there will be some mixing of the sample and mobile phase, effectively raising the acetonitrile content of the sample at the column inlet. Another approach is to inject the aqueous sample "sandwiched" between two small plugs of acetonitrile in the injector needle. In this case, a small portion (typically on the order of the volume of sample itself) of pure acetonitrile is drawn into the needle, then the sample itself, and then another small portion of acetonitrile (11). Here, again, these fluid will mix at the column inlet, effectively raising the acetonitrile content of the sample as it begins moving into the column. In our own work, we have found this approach to be effective for separations of monoclonal antibodies.
Summary
Many of the challenges we face in troubleshooting modern LC methods are as old as LC technology itself. However, as separation technologies evolve (for example, with the increase in use of HILIC separations), new troubleshooting challenges emerge, too. In this column, I have discussed scenarios in which problems can be related to the quality of solvents and reagents used in LC methods, as well as specific problems related to the use of highly aqueous sample solvents in HILIC separations. This is a great example of a situation where deeper understanding of the behavior of the method itself can help us be more effective in the process of LC troubleshooting.
References
(1) D.R. Stoll, LCGC North Am.36, 498–504 (2018).
(2) D.R. Stoll, LCGC North Am.35, 434–439 (2017).
(3) B.J. VanMiddlesworth and J.G. Dorsey, J. Chromatogr. A1236 77–89 (2012). doi:10.1016/j.chroma.2012.02.075.
(4) D.R. Stoll, R.W. Sajulga, B.N. Voigt, E.J. Larson, L.N. Jeong, and S.C. Rutan, J. Chromatogr. A1523, 162–172. (2017) doi:10.1016/j.chroma.2017.07.041.
(5) J. Layne, T. Farcas, I. Rustamov, and F. Ahmed, J. Chromatogr. A 913, 233–242 (2001). doi:10.1016/S0021-9673(00)01199-7.
(6) I. Bažkirova, G. Ruženaite, V. Olžauskaite, and A. Padarauskas, Chemija28, 233–239 (2017).
(7) J. Ruta, S. Rudaz, D.V. McCalley, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A1217, 8230–8240 (2010). doi:10.1016/j.chroma.2010.10.106.
(8) B. Chauve, D. Guillarme, P. Cléon, and J.-L. Veuthey, J. Sep. Sci. 33, 52–764 (2010). doi:10.1002/jssc.200900749.
(9) J.C. Heaton and D.V. McCalley, J. Chromatogr. A1427, 37–44 (2016).. doi:10.1016/j.chroma.2015.10.056.
(10) A. Periat, S. Fekete, A. Cusumano, J.-L. Veuthey, A. Beck, M. Lauber, and D. Guillarme, J. Chromatogr. A 1448, 81–92 (2016). doi:10.1016/j.chroma.2016.04.056.
(11) D.R. Stoll, D.C. Harmes, G.O. Staples, O.G. Potter, C.T. Dammann, D. Guillarme, and A. Beck, Anal. Chem. 90, 5923–5929 (2018). doi:10.1021/acs.analchem.8b00776.
ABOUT THE COLUMN EDITOR

Dwight R. Stoll

Dwight R. Stoll is the editor of "LC Troubleshooting." Stoll is a professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D–LC for both targeted and untargeted analyses. He has authored or coauthored more than 50 peer-reviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC's editorial advisory board. Direct correspondence to: LCGCedit@ubm.com










Mobile Phase Buffers in Liquid Chromatography: A Review of Essential Ideas
December 11th 2024In this installment of "LC Troubleshooting," Dwight Stoll discusses several essential principles related to when and why buffers are important, as well as practical factors, such as commonly used buffering agents, that are recommended for use with different types of detectors.


In-Loop Analyte Degradation in Two-Dimensional Liquid Chromatography: Example and Solutions
September 13th 2024In this installment of “LC Troubleshooting,” we describe an artifact that can arise because of compound degradation during the transfer of fractions of the first dimension (1D) column effluent to the 2D separation.


Eyes on the Prize: Overcoming Uncertainty to Realize the Power of 2D-LC Separations
June 10th 2024In this month’s column, I highlight some of the primary considerations we face in method development and point to resources that can help users overcome uncertainty and develop highly effective 2D-LC methods.

