Volatile Separation: A Low-Density Approach
Separating and quantifying both volatile and nonvolatile compounds in complex mixtures is a costly and time-consuming process presenting significant technical challenges. Fabrice Gritti from the Instrument, Core Research, Fundamental Group at Waters Corporation discusses his team’s unique solution to this problem, combining high-vacuum technology and low-density fluid chromatography (LDFC) with carbon dioxide as a mobile phase for a rapid and complete baseline separation of both volatile and nonvolatile compounds on a single instrument, single column, and a single run without the associated loss of resolution.


Photo Credit: Ilya Chalyuk / Nadezhda Molkentin/Shutterstock.com

Separating and quantifying both volatile and nonvolatile compounds in complex mixtures is a costly and time-consuming process presenting significant technical challenges. Fabrice Gritti from the Instrument, Core Research, Fundamental Group at Waters Corporation discusses his team’s unique solution to this problem, combining high-vacuum technology and low-density fluid chromatography (LDFC) with carbon dioxide as a mobile phase for a rapid and complete baseline separation of both volatile and nonvolatile compounds on a single instrument, single column, and a single run without the associated loss of resolution. - Interview by Lewis Botcherby
Q. Analyzing volatile compounds in complex mixtures is a costly and timeâconsuming process. Why is this so? And has it adversely affected any important areas of research?
Fabrice Gritti: The separation and quantification of volatile compounds present in complex mixtures is a technically challenging and costly process because their physical properties are different from those of the heavier components: their molecular weight is usually small and they have a high vapour pressure, so their boiling point at room temperature is very low. As a single class of compounds, they can easily be separated by gas chromatography (GC), which uses inert mobile phases (helium or nitrogen). Their separation can be achieved along coated capillary or packed columns using either isothermal or temperatureâprogrammed conditions, which control the partition coefficient of the volatile compounds between the liquid stationary phase (polydimethylsiloxane most often) and the gas phase. GC offers excellent selectivity for this class of compounds.
The main problem with more complex mixtures is that the less or nonvolatile fractions would remain stuck at the entrance of the GC column, even at temperatures as high as 400 °C. So, a second and more suitable separation technique involving stronger analyte–mobile phase interactions is needed: liquid chromatography (LC) or supercritical fluid chromatography (SFC) are two potential candidates. Adding one separation technique to complete the full analysis inevitably increases time and costs. This has motivated our research towards the development of a unique technique capable of analyzing such complex mixtures by using a single instrument (cost reduction), on a single column (1D chromatography), and during a single chromatographic run (time reduction).
LC alone could not be a viable solution because the interactions between the volatile compounds and the liquid phase are too strong: the totality of the volatile fraction present in the mixture would elute close to the hold-up time without any separation (see illustration of this separation problem in Figure 1 for terpenes present in a complex mixture).

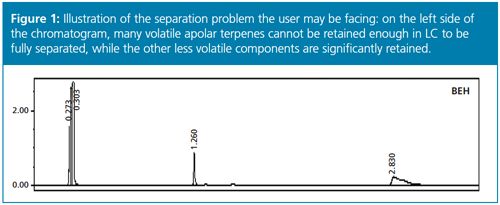
Therefore, a new strategy should be proposed and validated experimentally.
Q. In a recent publication, you explored low-density fluid chromatography (LDFC) as a potential solution to the difficulties of analyzing volatile compounds in complex mixtures (1). What challenges did you encounter when attempting to utilize this technique and how did you overcome them?
FG: The main challenge we are classically facing when trying to resolve simultaneously volatile and nonvolatile compounds is the lack of flexibility pertaining to the intensity of the
analyte–mobile phase interactions. It is either too weak (in GC) or too strong (in LC), regardless of the selected separation parameters (temperature, gradient).
This is where SFC and low-density fluid chromatography (LDFC) come into play. The well-known advantage of SFC over LC and GC mobile phases is that the experimenter can easily adjust its elution strength (or propensity to establish strong or weak interactions with the analytes) by controlling its temperature, pressure, and the amount of organic solvent added to it. For instance, on one end of the spectrum, at room temperature (298 K), high pressures (>1500 psi), and in the presence of methanol (up to 50% in volume), carbon dioxide–methanol eluent mixtures have densities similar to that of a liquid
(~1 g/cm3): they act as a liquid in terms of elution strength and are suitable for the elution of nonvolatile fractions. On the other end of the spectrum, take a much higher temperature of around 150 °C, a
low pressure around 1500 psi, in the absence of organic solvent, and the density of carbon dioxide falls to only 0.15 g/cm3, which is well suited for the retention and separation of the volatile fraction. LDFC using pure carbon dioxide as the mobile phase can then retain and solve the most volatile compounds present in a complex mixture.
One could now think that the resolution of volatile compounds could definitely be achieved by applying LDFC or SFC methods at high temperatures, low pressures, and in the absence of organic solvent. However, under conventional operating conditions (standard 2.1–4.6 mm × 15–25 cm analytical columns at flow rates of a few mL/min, pressure drops around 50 bar, and standard air-oven compartments), we are facing a known problem inherent to LDFC: low-density carbon dioxide is extremely expansible (its Joule-Thomson coefficient is very large), therefore, it cools down as it decompresses from the inlet to the outlet of the column. The cooling heat power can be as large as 50 Watt/m (negative thermal effects on column efficiency occur when this power exceeds only 5 Watt/m) and the eluent temperature may drop by 50 °C from the inlet to the outlet of the column. This means that the column may absorb a large amount of heat from its surrounding environment if it is not properly thermally insulated. Under steady-state temperature regime, a significant heat flux circulates from the column wall to the centre of the bed and generates radial temperature gradients across the column diameter. As a result, the density of carbon dioxide and the local migration velocity of the analytes are both larger in the wall than in the central regions of the bed, which causes a serious and nefarious deformation of the peak shapes. To summarize, LDFC using pure carbon dioxide successfully promotes retention of volatile compounds, yet this must be paid by a loss in resolution (see experimental illustration of the new problem in the top chromatogram shown in Figure 2).

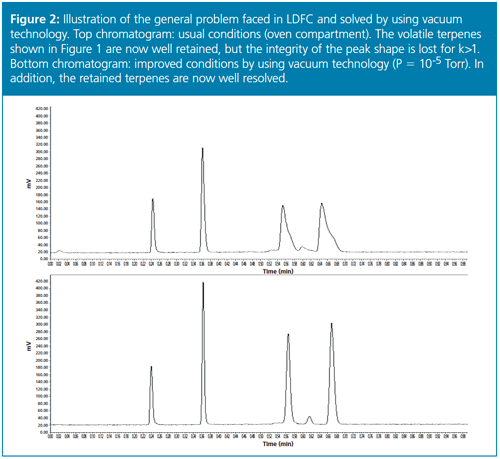
Therefore, improvements in LDFC must be developed to eliminate this inevitable and unacceptable peak deformation observed with low-density carbon dioxide. This is where vacuum technology in combination with LDFC came into play.
Q. The use of vacuum technology in combination with LDFC for terpenes and n-alkanes worked incredibly well (1). Were you surprised in any way by the results you obtained?
FG: Before performing any experiments in LDFC with pure carbon dioxide, the original belief was that by applying a high vacuum (< 10-5 Torr) around the column body, one would eliminate any sort of heat exchanges (by convection and diffusion) between the column and the external environment (air in the oven compartment or laboratory air) at the exception of the residual heat exchange by radiation (note that the stainless steel external surface area of the column was even covered with a thin aluminum tape to minimize heat exchange by radiation). The goal was to minimize the amplitude of the radial temperature gradients and to restore peak shape and the highest allowable column efficiency as predicted by the theory. Quite surprisingly, this idea happened to work extremely well in ultrahigh-pressure liquid chromatography (UHPLC), in which case the column releases large amounts of heat to the external environment (inversely, the column is absorbing some heat in LDFC). The average relative gain in column efficiencies typically went from +20 to +50% at the highest allowable speed, irrespective of the nature of the packing material considered (fully porous silica particles, fully porous hybrid organic or silica particles, and superficially porous particles).
What we have experimentally learned and demonstrated is that in UHPLC this relative gain in column efficiency was a result of the establishment of a more uniform temperature profile across the column internal diameter, for example, to a more uniform viscosity and flow velocity profile from the centre to the wall of the column. To summarize, the trans-column or longârange eddy dispersion HETP term in the van Deemter equation (the A term) was significantly reduced at high speeds. The effect was so intense that it was no longer possible to observe the minimum HETP at the highest allowable flow rate.
From these initial and very encouraging results observed in UHPLC, we naturally decided to extend this concept of applying a high-vacuum around the chromatographic column from UHPLC to LDFC. Indeed, the fundamental problem to circumvent was strictly the same: combat the existence of radial temperature gradients across the column internal diameter whether the heat is released (UHPLC) or absorbed (LDFC) by the column. Therefore, it was only half a surprise to observe an improvement in peak shape and resolution for volatile compounds by combining high vacuum technology with LDFC, at least when the retention factors were smaller than 5 (See the bottom chromatogram in Figure 2 in comparison to the top chromatogram recorded under standard conditions and Figure 3). However, I was personally and pleasantly surprised to observe that the undetectable peaks of semivolatile compounds (intermediate fractions between volatile and nonvolatile fractions) with retention factors as large as 25 could now become detectable in LDFC. Their peak shape was restored from severely spread to quasi-Gaussian and narrow bands in line with the expected efficiency of the column used. It is important to keep in mind that in LDFC the retention of strongly retained compounds is extremely sensitive to any variations of the density of carbon dioxide. Their peak shape is strongly affected by any minor change in the local density of the eluent across the column internal diameter. Hence, it is crucial to thermally insulate the body of the chromatographic column in LDFC for the sake of improving both resolution and sensitivity relative to conventional air ovens. We were also positively surprised to observe that vacuum technology was even more beneficial in LDFC than in UHPLC. The band deformation observed in LDFC is essentially due to the local change in retention of analyte across the column diameter, while, in UHPLC, it is mostly caused by the local change in the flow velocity.

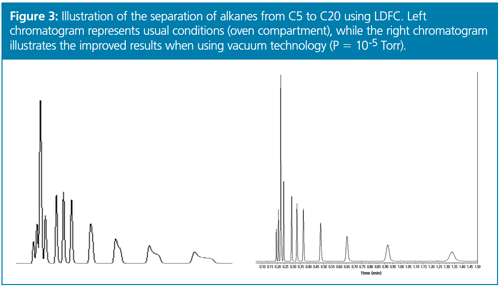
Now that LDFC can be successfully operated for the separation of volatile compounds, it is also very important to analyze properly on the same instrument and with the same column the less- and nonvolatile compounds present in the complex mixture. This can be done by lowering the eluent temperature, increasing the ABPR pressure, or increasing the amount of organic solvent in the carbon dioxide mobile phase. The transition from isothermal or isobaric LDFC to conventional SFC gradients for the separation of less or nonvolatile compounds should be conducted with great caution.
Q. Your work raised a fundamental issue relating to the application and performance of ABPR pressure gradients, suggesting a compromise must be found between the application of a high-vacuum and wall temperature-control in an oven compartment. What do you believe is the best compromise and why?
FG: That is right. The analysis of the less or nonvolatile compounds requires different operating conditions in terms of temperature, ABPR pressure, and organic solvent fraction relative to those applied for the separation of volatiles. Volatile compounds are best separated under isothermal (high temperature) and isobaric (low ABPR pressure) conditions. In contrast, ABPR pressure and organic solvent composition gradients are usually applied to elute the high-molecularâweight compounds present in the sample mixture. Inevitably, during such gradients, the physical state and physicoâchemical properties of the eluent change with increasing time (see an example in Figure 4).

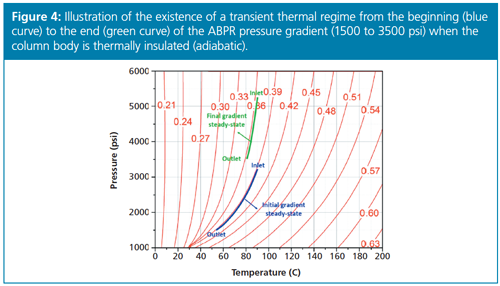
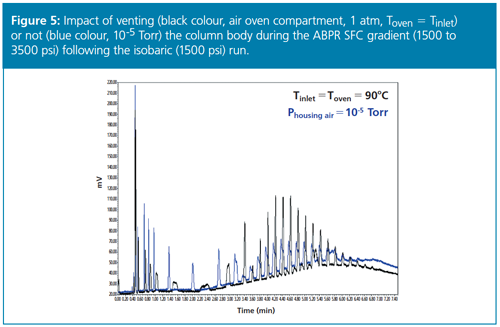
The large difference between the initial and final temperature-pressure states of the eluent induces a transient thermal regime inside the column (involving the column stainless steel hardware and the packing material) even though it is plunged into a high-vacuum chamber. Heat is redistributed between the eluent, the packed material, and the stainless steel column hardware. The negative consequence is the emergence of temporary radial temperature gradients across the bed while the gradient is running, which limits the resolution level of the separation (see Figure 5, blue colour signal). The question is then to find out 1) how one can mitigate this transient thermal regime inherent to SFC gradients, and 2) which is the best compromise between the first adiabatic–isobaric–isothermal run and the second gradient run.
First, there is no absolute solution to this problem and only a compromise must be found experimentally between the initial isobaric–isocratic run (volatiles) and the SFC gradient run (nonvolatiles). In addition, it is not a straightforward task for the common practitioner because it requires knowledge of the change in the fluid enthalpy because the ABPR or the content in organic solvent varies along the column during the SFC gradient. Most often, these data are not even available in the literature for carbon dioxide–methanol mixtures. So, the user is often doomed to a series of trial-and-error experiments.
Second, however, a semi-empirical rule of thumb involves selecting the lowest allowable eluent temperature (until the selectivity is lost) in LDFC under vacuum to minimize the temperature change from the column inlet to the column outlet (it is because the isenthalpic lines in the P-T diagram are getting steeper as T is decreasing). Then, the second semiâempirical rule comprises applying the smoothest possible ABPR or organic solvent gradients-as long as the analysis time remains within an acceptable range-to minimize the amplitude of the transient radial temperature gradients.
Finally, if the volatile fraction elutes much before (at least 15 min for a 3.0 × 150 mm column size) the nonvolatile fraction, the chromatographer may have the option of venting the column from high-vacuum to atmospheric pressure and of fixing the oven temperature at a temperature equal or slightly lower than the inlet temperature. In so doing, during the whole SFC gradient, the inlet-toâoutlet cooling of the eluent remains moderate and the heat exchanged between the column wall and the oven is kept to a minimum. Performance is then improved (see Figure 5).

Q. What advice would you give to someone who wanted to use this technique?
FG: To summarize, anyone who is interested in analyzing volatile or semivolatile compounds by LDFC using carbon
dioxide as the mobile phase should no longer worry about the usual and serious loss of resolution when increasing temperature or decreasing the ABPR pressure. This can be prevented by application of a highâvacuum in the immediate surroundings of the column body. The full baseline separation of pentane and n-hexane can now be easily achieved with carbon dioxide in 20 s at an inlet temperature of only 90 °C, an ABPR pressure of 1500 psi, and a standard 15âcm long column packed with sub-2âµm silica-C18 particles. Performing similar analyses (see Figures 3 and 5) but
at much higher temperatures, such as in
the range 150–200 °C, would even allow the separation of more volatile compounds than pentane-provided the stationary phase could remain thermally stable.
Finally, the analysis of the less volatile fractions present in the sample mixture can still be run in series with the same instrument, the same column, and at the same flow rate from standard SFC gradient methods. The column oven compartment is vented back from highâvacuum to 1 atm, the oven temperature is adjusted to the desired temperature in conventional SFC gradients, and the inlet eluent temperature is kept at about 10 °C above the oven temperature to generate maximum peak capacity.
Reference
- Gritti et al., J. Chroma. A1472, 107–116 (2016).


Fabrice Gritti is currently working as a Principal Research Scientist at Waters Corporation in the Instrument, Core Research, Fundamental Group. Over the last 15 years, his research interests have involved liquid and solid adsorption thermodynamics and mass transfer in heterogeneous media for characterization and design optimization of new HPLC column and instrument technologies. He has published over 240 peer-reviewed scientific articles. He was the recipient of the 2013 Chromatographic Society Jubilee Medal for his important contribution to the development of chromatographic science.
E-mail:Fabrice_Gritti@waters.com


















