Tips & Tricks GPC/SEC: Protein Analysis with Size-Exclusion Chromatography
The Column
Gel permeation chromatography/size-exclusion chromatography (GPC/SEC) is the standard method to separate samples by molecular size. In protein analysis, size-exclusion chromatography is either applied to detect and quantify aggregation, or to measure the complete molar mass distribution. However, method development is not trivial and the choice of suitable detection options is crucial.


Photo Credit: -strizh-/Shutterstock.com

Daniela Held and Thorsten Hofe, PSS Polymer Standards Service GmbH, Mainz, Germany
Gel permeation chromatography/size-exclusion chromatography (GPC/SEC) is the standard method to separate samples by molecular size. In protein analysis, size-exclusion chromatography is either applied to detect and quantify aggregation, or to measure the complete molar mass distribution. However, method development is not trivial and the choice of suitable detection options is crucial.
In contrast to synthetic macromolecules and the majority of biopolymers, most proteins do not exhibit a molar mass distribution (1) but are uniform in molar mass. Nevertheless, fractionating techniques are useful for such monodisperse protein samples because they can identify or monitor potential aggregation. Traditionally sizeâexclusion chromatography (SEC) can be used to separate stable aggregates from monomers or fragments and, in combination with advanced detection such as light scattering (LS) or mass spectrometry (MS), also to determine their molar mass and size. For protein mixtures with a molar mass distribution (such as gelatins), gel permeation chromatography (GPC)/SEC is applied because it measures the complete molar mass distribution and gives access to a variety of quality (and safety) related results. A major challenge for GPC/SEC protein analysis is the development of a robust method that eliminates undesired interactions with the stationary phase present in the separation column.
What are the Advantages and Limitations of GPC/SEC?
A major advantage of GPC/SEC is that it is a nondestructive separation technique. It fractionates samples based on the sizes present. GPC/SEC is ideal for reducing the complexity of a sample before applying advanced detection techniques such as MS, LS, or viscometry. The reduction in complexity eases data evaluation and interpretation when compared to direct analysis of nonfractionated heterogeneous samples. Advanced detection can very often be used as an on-line method. If hyphenation is not applicable, GPC/SEC allows the sample fractions collected to be used for further characterization.
In GPC/SEC proteins can be analyzed under native conditions in solution, which is favourable as conformations and protein–protein interactions remain intact. Many GPC/SEC separations therefore preserve the biological activities of the macromolecules.
On the downside, the conditions for true GPC/SEC separations-those only based on the size of the protein in solution-can be cumbersome to achieve. Interactions between the sample and the stationary phase are often encountered, resulting in unexpected peak shapes, unstable retention times, and poor recovery. As different proteins exhibit different shapes (for example, globular-, rod-like-, or flexible chains), their sizes in solution often do not correlate directly with molar mass, thereby facilitating the need for more advanced detection methods for molar masses.
A thorough method development for protein analysis should comprise of selecting the right column (or column combination) with the matching stationary phase, particle size, and porosity, adjusting the pH and ionic strength of the mobile phase, and selecting the best detection option for a series of samples. An essential part is also evaluating the chromatographic recovery of all components and aggregates present in the sample.
Pitfalls in Method Development
Proteins need to be analyzed in aqueous solutions and are therefore a subcategory of aqueous GPC/SEC. All general method development tips for GPC/SEC are applicable to protein method development as well. The influence of column dimensions, particle size, temperature, flow rate, and injected mass have been described in several GPC/SEC Tips & Tricks instalments (2–5). This article will therefore focus on the special requirements for proteins only.
One advantage of proteins over synthetic polymers or large biomolecules is that proteins are usually small and the size distribution within a sample is relatively narrow. The chromatographic benefits of small particles can therefore be fully exploited. In addition, the narrow distribution means that columns with very flat calibration curves (high resolution in a narrow molar mass range) can be used. Silica-based GPC/SEC columns are therefore still preferred for most protein applications.
A disadvantage of proteins from a chromatographic point of view is that they have a large number of functional or charged groups and may possess larger hydrophobic parts than other waterâsoluble macromolecules. These two characteristics make it difficult to develop a true GPC/SEC method with no (or at least minimized) electrostatic and hydrophobic interaction between the column stationary phase and the protein. Interaction can result in adsorption (reduced recovery, often no peaks detectable), shifted elution times (higher or lower elution volumes), or distorted peak shapes (tailing).
To avoid or reduce interactions, the pH value, the ionic strength or salt content, and the use of potential organic modifiers need to be adjusted. However, the concentration of the salts or the modifier added is limited because it also influences the solubility of the sample. If it is not possible to develop a stable GPC/SEC method a change of stationary phase (from the silica column to a polymerâbased column) should be attempted.
A good starting point for method development is running the protein at its isoelectric point (pI). The pI of a protein is the pH value at which positive and negative charges resulting from the different amino acids comprising the protein are balanced. Neither the positively charged ammonium nor the negatively charged carboxyl groups are dominating here. An ideal GPC/SEC method for every single protein would be run with an aqueous solution with a pH matching the pI.
To avoid the cumbersome approach of having a method for every single protein, the pH of the mobile phases is chosen to be close to pI and additional polyelectrolytes–monovalent salts, such as sodium chloride or potassium chloride, are added. The electrolyte will help to shield residual charges and reduce the undesired interaction between the proteins and the stationary phase.
Another parameter to be considered is the hydrophobicity of the protein. The polarity of the different R groups in the protein is important. Based on the R group an amino acid can be classified as charged, hydrophilic, or hydrophobic. Proteins containing many hydrophobic amino acids, such as leucine, alanine, and valine, are rather hydrophobic and cannot be analyzed under the same conditions as hydrophilic or charged proteins containing amino acids, such as serine, arginine, or histidine.
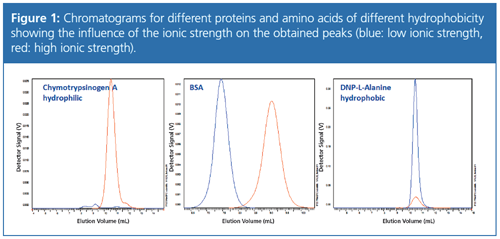
Figure 1 compares the influence of the ionic strength on the chromatograms of proteins with different hydrophobicity. A hydrophilic protein such as chymotrypsinogen A should be measured at high ionic strength; bovine serum albumin (BSA), a protein that is neither hydrophobic nor hydrophilic, can be measured at high and low ionic strengths; and hydrophobic amino acids such as alanine or substituted alanines should be analyzed at low ionic strengths. If the ionic strength is adjusted according to the polarity of the protein the separation will fail.
Finally, it should be mentioned that unexpected peak shapes (for example, with tailing) have been shown to be the result of using standard liquid chromatography (LC) equipment and detector cells. To reduce metal–protein adducts or undesired protein interactions it might be required to use bioâinert or biocompatible chromatographic systems and column hardware. Such systems are also of advantage when harsh conditions are applied, such as extreme pHâvalues or high salt content. Bacteriocides or bacteriostats can help to overcome bacterial contamination. Without this algae growth can occur in aqueous mobile phases within a few hours and may cause severe problems.

Detection Options
Proteins can be detected using UV detectors. This is highly convenient because UV detectors are easy to use, linear in response over a wide range, and sensitive and baseline stable. Each UV wavelength range has its advantages: 280 nm is very often selected as a standard wavelength. Near UV or longer wavelengths (380–315 nm) detect aromatic amino acids, such as tryptophan. Higher sensitivity is provided by selecting a wavelength in the far UV range (280–200 nm) at low wavelengths such as 220 nm, where the amide peptide bond has a strong absorbance. There are also many setups where two different wavelengths are recorded simultaneously to measure the protein concentration. The combination of this dual-wavelength detection approach has been proposed for purity investigations, where the lower wavelength provides the sensitivity for the low abundant species, while the higher wavelength provides a higher linear range for the major species (the monomer, for example) (6).
To overcome the limitation of sizeâbased GPC/SEC separations, either MS or LS detection can be used to determine the molar mass directly. Both methods should be understood as complementary techniques because they offer solutions in completely different application fields. MS is very strong when it is required to determine very specific details about a limited number of individual species of not too high molar mass. MS is by far the most precise and accurate way to determine molar masses. However, when sample complexity and molar masses increase, the density of the resulting mass spectrum makes it very often impossible to interpret the data and assign the results. Fortunately, in these cases LS can be used because this technique is well suited for analyzing polydisperse high molar mass samples. Another advantage is that LS is very sensitive to high molar masses, even at low concentrations. LS can often detect higher aggregates with higher sensitivity than any other detector.
The greatest challenge when hyphenating GPC/SEC with MS is the composition of the required mobile phase. Mobile phases in GPC/SEC often contain high concentrations of nonvolatile, MS-incompatible salts, which can lead to problems with contamination of the mass spectrometer and ion suppression. A hyphenated approach is therefore much easier to achieve with LS. There is no need to evaporate the mobile phase. An additional advantage is that for LS detection, structure information is still available, which would be lost with MS detection when the proteins are ionized and vaporized.
There are many LS detectors on the market and they mainly differ by the number of angles that simultaneously acquire the scattering intensities (7). The majority of globular proteins are so small in dimension that 90° light scattering is fully sufficient to measure the molar mass. Unfortunately, this also means that it is not possible to measure the size (radius of gyration) with multi-angle light scattering detection (MALLS) for many proteins. Other techniques, such as dynamic light scattering (DLS, also known as QUELS) for measuring the hydrodynamic radius (Rh), must be applied.
The most common detector in GPC/SEC for synthetic polymers, the refractive index detector (RI), is also used for protein analysis. However, an RI detector is mainly used in combination with a LS detector because it can determine the refractive index increment, dn/dc, on-line. This sample-related parameter strongly influences the accuracy of light scattering results. It depends on many experimental settings including the LS detector wavelength and the solvent for example. Often an averaged dn/dc of 0.185 mL/g is applied for all proteins, however, care should be taken here because the dn/dc can vary significantly with the protein type (8).
All other GPC/SEC detection options can be used. Some proteins can be analyzed with fluorescence detectors with an improved sensitivity and selectivity.
Viscometers are becoming more and more common to distinguish between denaturation and aggregation. They can detect density differences and help to identify structure changes. The multidetection approach is already common for the GPC/SEC analysis of synthetic polymers, and is becoming more popular for protein analysis.
How Do the Results Look?
Figure 2 shows the raw data and results for a UV-LS setup used to investigate protein aggregation. The combined analysis of UV trace and light scattering trace gives direct access to the molar masses of dimer and monomer without column calibration. As the obtained molar mass of the early eluting smaller peak is approximately twice the molar mass of the later eluting larger peak, this is an indication of dimerization. Analyzing the peak areas of the UV trace allows the dimer content to be determined to around 10%.

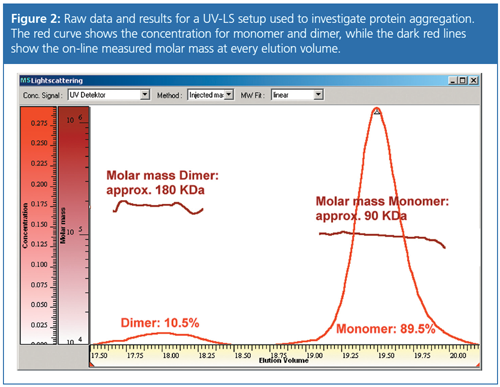
Figure 3 shows an overlay of the chromatograms obtained for a full length monoclonal antibody (mAb) and antibody fragments analyzed on the same set of columns. The red curve shows the UV signal of the full length antibody and its dimers, while the blue curve shows the UV signal of antibody fragments and their high level aggregates.

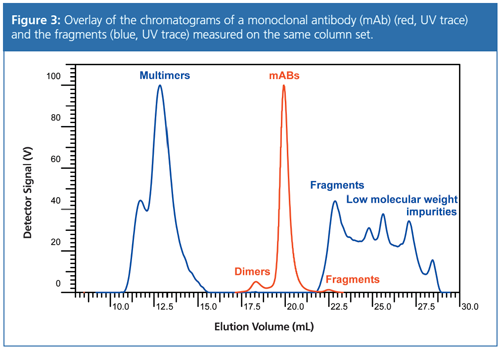
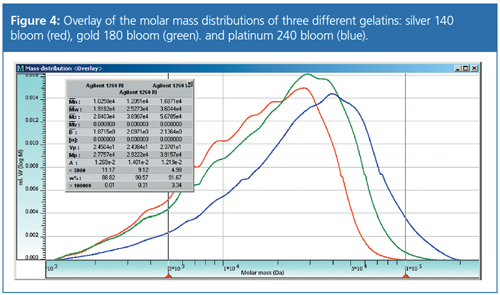
Figure 4 shows the results for a protein exhibiting a molar mass distribution. In the overlay three different gelatins can be clearly differentiated, the molar mass averages and the molar mass distribution can be easily determined.

Summary
- GPC/SEC is a powerful technique to investigate protein aggregation. It can separate proteins under native conditions based on their sizes in solution.
- Size-based separation conditions typically require mobile phases with adjusted ionic strengths. Method development should also include recovery investigations.
- Advanced detection options (MS or LS as complementary techniques) can help to overcome GPC/SEC limitations and measure the molar masses directly.
- Other typical GPC/SEC detectors, such as RI or a viscometer, can provide dn/dc (required for LS evaluation) or structural information.
References
- D. Held, The Column3(8), 23–24 (2007).
- D. Held, The Column9(6), 9–12 (2013).
- D. Held, The Column8(22), 8–11 (2012).
- D. Held, The Column7(2), 14–16 (2011).
- T. Hofe and D. Held, The Column 5(6), 9–12 (2009).
- M.D. Bond, M.E. Panek, Z. Zhang, et al., J. Pharm. Sci.99(6), 2582–97 (2010).
- D. Held and P. Kilz, The Column5(4), 28–32 (2009).
- H. Zhao, P.H. Brown, and P. Schuck, Biophys. J.100(9), 2309–17 (2011).
Daniela Held studied polymer chemistry in Mainz, Germany, and works in the PSS software and instrument department. She is also responsible for education and customer training.
Thorsten Hofe studied chemistry in Mainz, Germany, and Toronto, Canada. He is the head of the PSS production department (columns, reference material, and polymer synthesis). He is also involved in education and customer training.
E-mail:DHeld@pss-polymer.comWebsite:www.pss-polymer.com
















, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

