The Use of HILIC Zwitterionic Phase Superficially Porous Particles for Metabolomics Analysis
Special Issues
This article highlights the use of a new HILIC zwitterionic phase on superficially porous particles. A study on the use of a novel mobile-phase additive to achieve superior peak shape and isomer separation is also discussed, as well as improved LC–MS detection capabilities for metabolomics analysis.
Recently, superficially porous particles (SPPs) have generated significant interest because of the enhanced separation efficiency achieved at lower back pressure compared to that obtained using fully porous particles of the same particle size. Although many reversed-phase chemistries are available on SPPs, the chemistries for hydrophilic-interaction chromatography (HILIC) are limited. This article highlights the use of a new HILIC zwitterionic phase on superficially porous particles. A study on the use of a novel mobile-phase additive to achieve superior peak shape and isomer separation is also discussed, as well as improved liquid chromatography–mass spectrometry (LC–MS) detection capabilities for metabolomics analysis.
Reversed-phase chromatography is the most popular high performance liquid chromatography (HPLC) method for the purification and analysis of a wide variety of analytes (1). However, analysis of polar metabolites such as amino acids, nucleotides, and organic acids by reversed-phase chemistries can be difficult because of low retention by the stationary phase (2). To promote the retention of polar analytes on reversed-phase columns, derivatization methods or ion-pairing reagents can be used (3,4), but there are disadvantages of these approaches that have been described previously (5,6). In contrast, hydrophilic-interaction chromatography (HILIC) serves as an alternative approach to analyze hydrophilic and polar analytes (7,8). Here, we developed a new HILIC chemistry on superficially porous particles (SPPs) that withstands high-pH mobile-phase solvents and chromatographically separates underivatized carbohydrates, amino acids, and metabolites. The wide operating pH range of the new HILIC column allows chromatographers to test a larger pH range on a single column to determine the optimal chromatographic performance for their targeted analytes. Moreover, high-pH mobile-phase solvents can limit the interactions between stainless steel surfaces and phosphorylated analytes (9), which is thought to be a major contributor to the difficulties associated with the analysis of phosphorylated and carboxylated metabolites (10,11). Furthermore, a detailed study carried out to investigate the effect of bioinert hardware and a novel mobile-phase additive that deactivates metals in the sample flow path are presented.


Experimental
Column Stability Study
For the high-pH test, solvent A was made by mixing 125 mL of 30% ammonium hydroxide in 875 mL of water. The pH was measured at around 11 in aqueous buffer. Solvent B was made by adding 4 L of acetonitrile into the mobile-phase container. HILIC was performed using a 100 mm × 2.1 mm Poroshell 120 HILIC-Z column (Agilent Technologies). The flow rate was 0.40 mL/min and column temperature was set at 30 °C. A sample volume of 1.0 µL was injected onto the column every 80 column volume push at 40:60 (v/v) A:B. The settings for evaporative light scattering detection (ELSD) were 60 °C, 3.5 bar, and 30 Hz. For the high-temperature test, the flow rate was 0.40 mL/min and the column temperature was set at 80 °C. A sample volume of 1.0 µL was injected onto the column for every 80 column volume push at 10:90 (v/v) 100 mM ammonium acetate at pH 7.0 acetonitrile. The settings for ELSD were 60 °C, 3.5 bar, and 30 Hz.
Liquid Chromatography–Mass Spectrometry (LC–MS) Analysis of Amino Acids
Amino acid analysis was performed using a 100 mm × 2.1 mm Poroshell 120 HILIC-Z column (Agilent Technologies). First, a stock solution of 200 mM ammonium formate (adjusted to pH 3.0 with formic acid) in water was prepared. Solvent A was made by mixing 100 mL of the stock solution and 900 mL of water. Solvent B was made by mixing 100 mL of the stock solution with 900 mL of acetonitrile, resulting in mobile-phase A and B both having an ionic concentration of 20 mM. The flow rate was 0.80 mL/min and the column temperature was set at 30 °C. An 18 amino acid sample mixture in 0.1 M hydrochloric acid (2.5 mM amino acids, except cysteine at 1.25 mM) was diluted 1000-fold with starting condition mobile phase, and 0.10 µL of sample volume was injected onto the column for each experiment. The gradient elution profile was from 100% to 70% B for 12 min and the column was equilibrated with 100% B for 4 min before subsequent analysis. A multiple reaction monitoring (MRM) method was set up to acquire data on a 6470 QQQ system (Agilent Technologies).
LC–MS Analysis of Metabolites
Stock solutions of the analytes were made in Milli-Q purified water at 5 mg/mL. Sample solutions were made by diluting the stock to 1 ng/µL (ppm) in 80:20 acetonitrile–water. HILIC was performed using a 50 mm or 150 mm × 2.1 mm Poroshell 120 HILIC-Z column in both stainless steel and PEEK-lined stainless steel hardware (Agilent Technologies). Stock solutions of 100 mM ammonium acetate (adjusted to pH 9.0 with ammonium hydroxide) in water were first made. Solvent A was prepared by mixing 100 mL of the stock solution and 900 mL of water, which yields a final concentration of 10 mM ammonium acetate (pH 9.0) in water. Solvent B was made by mixing 100 mL of the stock solution with 900 mL of acetonitrile, which yields a final concentration of 10 mM ammonium acetate (pH 9.0) in 90% acetonitrile. A deactivator additive (Agilent Technologies) was spiked into indicated solvents at a final 5 µM concentration for analysis. The flow rate was 0.25 mL/min and the column temperature was set at 25 °C. A sample volume of 0.2–3 µL was injected onto the column for each experiment. After loading of the sample solution, the column was conditioned with 90% solvent B for 2 min before the gradient with solvent A was applied. The gradient elution profile was from 90% to 60% B for 10 min followed by washing with 60% B for 3 min. The column was equilibrated with 90% B for 8 min before subsequent analysis. Full MS (MS1) data were acquired with a mass range of 50–1000 m/z and an acquisition rate of 1 spectrum/s on a 6545 Q-TOF system (Agilent Technologies). An MRM method was also set up to acquire data on a 6490 iFunnel QQQ system (Agilent Technologies). The instruments were operated in negative mode for metabolite analysis except for polyamines, which was analyzed in positive mode.
Cell Culture Study
K562 leukemia cells were cultured in suspension in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum. A portion of the cells and media collected immediately (day 0) or six days later (day 6) were centrifuged at 250g for 5 min to pellet the cells. The collected growth media (100 µL) was mixed with 400 µL of 50% acetonitrile and centrifuged at 10,000g for 5 min. A 0.5-µL volume of the supernatant was subjected to HILIC–LC–MS analysis.
Results and Discussion
Stability at High pH or High Temperature for Carbohydrates
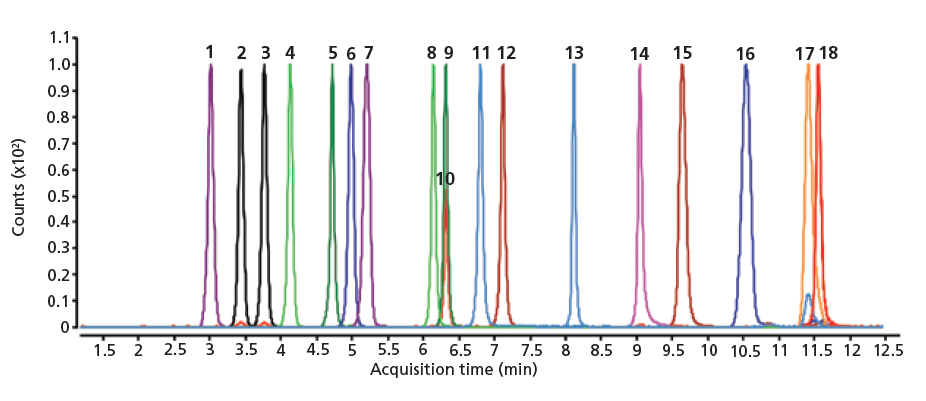
The 2.7-µm Poroshell 120 HILIC-Z particles synthesized with a proprietary hybrid protected zwitterionic bonding are resistant to silica dissolution at high-pH conditions. The lifetime of the HILIC-Z column was tested under conditions suitable for carbohydrate analysis, wherein high-pH or high-temperature methods are commonly used to reduce double peaks from anomeric configurations. It was previously reported that several HILIC columns were highly unstable and even seriously damaged after exposure to basic condition (pH >10) (12). As shown in Figure 1a, the retention time of raffinose remained stable after passing over 6000 column volumes of high-pH mobile-phase buffer (pH 11) in 60% acetonitrile through the column at 30 °C. In contrast, although the combination of neutral pH and 80 °C offers faster separations and lower back pressure, this approach resulted in noticeable retention time loss for the carbohydrates (Figure 1b). The retention time loss indicated degradation of column chemistry under high-temperature analytical conditions. Nevertheless, the retention time of most sugars dropped less than 10% over 10,000 column volumes, while narrow peak widths and peak shapes were maintained.

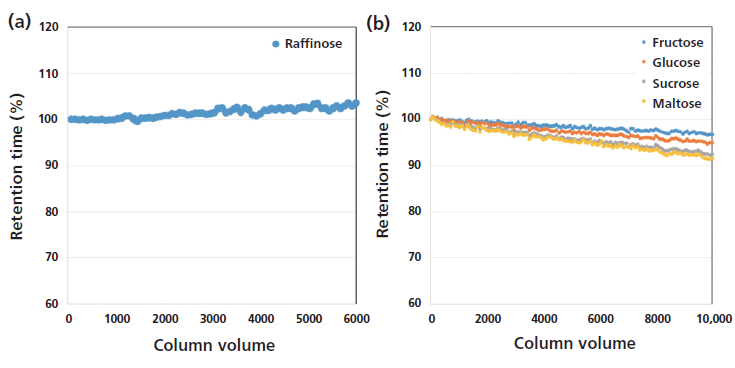
Figure 1: Lifetime analysis with sugars were conducted at (a) high pH and (b) high temperature. Methods parameters are (a) 60:40 acetonitrile–3.75% ammonium hydroxide at pH 11, 30 °C (b) 90:10 acetonitrile–100 mM ammonium acetate at pH 7.0, 80 °C; 0.4 mL/min; (a,b) ELSD: 60 °C, 3.5 bar.
Separation of Underivatized Amino Acids
Historically, amino acids are analyzed by gas chromatography (GC) or cation-exchange or reversed-phase LC with ultraviolet (UV) or MS detection. However, the use of derivatization agents is not optimal for LC–MS because it often reduces MS sensitivity and adds complexity to the spectra. Although the polarity of amino acids makes analysis by reversed-phase LC difficult, it makes them a perfect candidate for analysis with the combination of HILIC coupled to MS.
The separation of underivatized amino acids using the HILIC column with low-pH mobile-phase solvents and MS detection in positive analysis mode resulted in the best MS sensitivity and chromatographic performance. As shown in Figure 2, the HILIC column separated a mixture of 18 underivatized amino acids in 12 min while providing excellent peak shape and resolution. This analysis includes the complete separation of leucine and isoleucine isomers under LC–MS-friendly conditions.

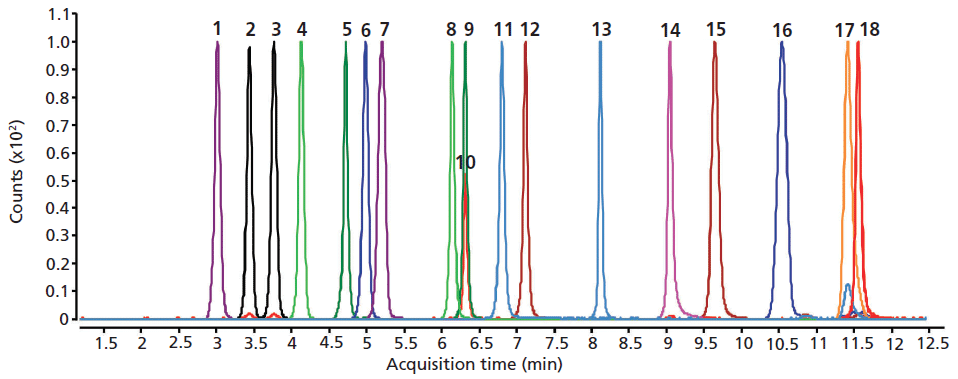
Figure 2: LC–MS analysis of 18 underivatized amino acid mixture on a 100 mm × 2.1 mm HILIC-Z column. Peaks: 1 = Phe, 2 = Leu, 3 = Ile, 4 = Met, 5 = Tyr, 6 = Val, 7 = Pro, 8 = Ala, 9 = Thr, 10 = Cys, 11 = Gly, 12 = Ser, 13 = Glu, 14 = Asp, 15 = His, 16 = Arg, 17 = Lys, 18 = 2-Cys (dimer).
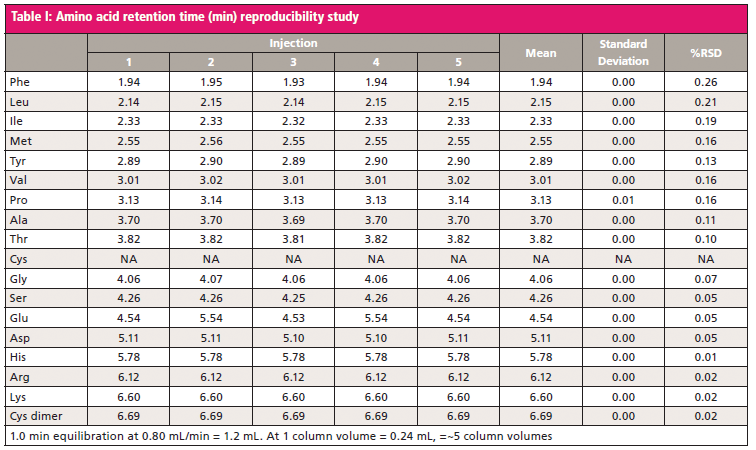
The perceived limitations of HILIC are extremely long column reequilibration times and inconsistent retention time reproducibility, which prevent extensive use of the technique. However, it has been demonstrated that a consistent reequilibration time results in reproducible analyte retention times; furthermore, analyte retention time variability is most often observed with extended reequilibration times with basic analytes on a bare silica stationary phase (13). With the HILIC column and a reequilibration of five column volumes, we achieved consistent amino acid retention times (Table I).

Effect of Mobile-Phase pH on Chromatographic Performance
A distinct advantage of the HILIC particles is the stationary phases's ability to withstand a wide operating pH range (pH 3–11) (Figure 1). In general, acidic mobile phases favor positive mode ionization, whereas basic mobile phases favor negative mode ionization (14). The wide operating pH feature was useful for resolving difficult to separate structural isomers such as citrate and isocitrate. As shown in Figure 3, citrate and isocitrate are coeluted when analyzed with mobile-phase buffers at pH 9.0. However, when the pH of the mobile-phase solvent was acidified to pH 6.8, citrate and isocitrate were resolved.

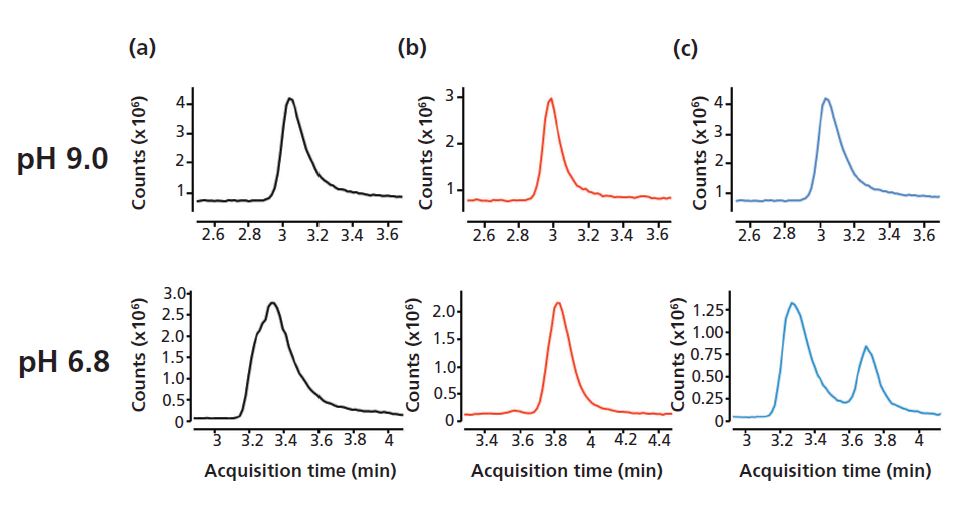
Figure 3: Citrate and isocitrate isomers were resolved using neutral-pH mobile-phase buffer: (a) citrate, (b) isocitrate, (c) citrate/isocitrate. Samples were analyzed with ammonium acetate mobile phase buffers at the indicated pH values using an 80–70% B gradient in 10 min.
The versatility of the stationary phases's operating pH also allows a wide range of metabolites to be evaluated. For example, polyamines such as putrescine, spermidine, and spermine were found to yield a better signal and peak shape at lower pH (3.5) compared to neutral pH (pH 6.8) conditions (Figure 4a). In contrast, phosphorylated nucleotides such as adenosine monophosphate (AMP), adenosine diphosphate (ADP), and adenosine triphosphate (ATP) were found to have higher signal and better peak shape under high-pH (9.0) conditions compared to lower-pH (3.5) conditions (Figure 4b). The nucleotide results are supported by a previously published report that basic solutions (pH >8.5) limit interactions between phosphorylated analytes and metal, which often coats the inner surface of LC columns (9). Choosing the best mobile-phase buffer conditions will undoubtedly be the first critical decision to yield optimal chromatography results for users.

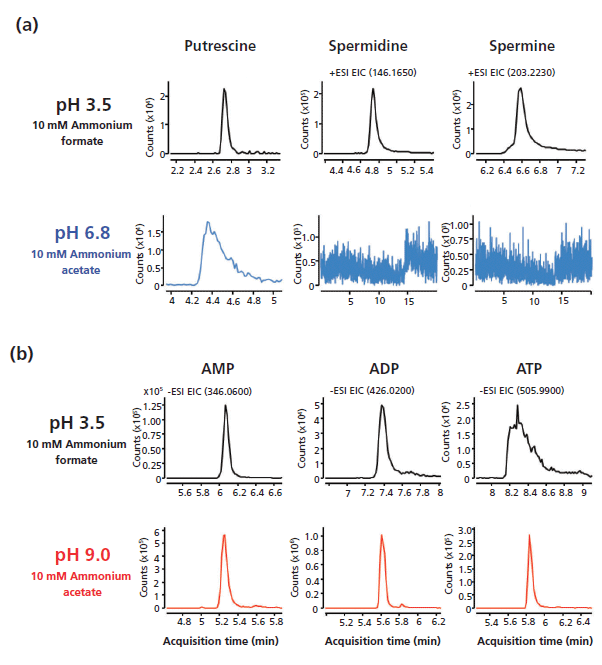
Figure 4: (a) Polyamines and (b) phosphorylated nucleotides were analyzed with mobile-phase solvents at the indicated pH values to determine the optimal analytical conditions.
Mobile-Phase Additives Enhance the Chromatographic Performance of Metal-Sensitive Metabolites
Severe peak tailing of phosphorylated and carboxylated metabolites is a well-documented issue in LC–MS analysis (10,11). The poor peak shapes are thought to be influenced by trace metal leaching from the chromatographic hardware or direct interaction of the analytes with metal oxides along the sample flow path (10). Metal chelators, such as ethylenediaminetetraacetic acid (EDTA), have previously been used to deactivate the LC system, or have been spiked into mobile-phase solvents or samples to improve the chromatographic performance of metal-sensitive analytes (10,11). However, these metal chelators are highly ionizable and cause ion suppression of target analytes (11). Thus, we sought to identify a mobile-phase additive that would be as effective at chelating metal ions as EDTA, but without the ion suppression effects. Here, we discovered a novel mobile-phase additive that could significantly improve the peak shape and signal strength of metal-sensitive metabolites (Figure 5). Moreover, switching column hardware from stainless steel to PEEK-lined improved ATP and malate's peak shape and signal intensity (Figure 5). Because of the high-pH tolerance of the HILIC column (Figure 1), analyzing the phosphorylated compounds under high-pH conditions also further limits the metal–analyte interactions (9,15). Thus, the combined use of the HILIC stationary phase in PEEK-lined column hardware, the high-pH mobile phase, and the deactivator additive yields excellent chromatographic performance for metal-sensitive analytes.

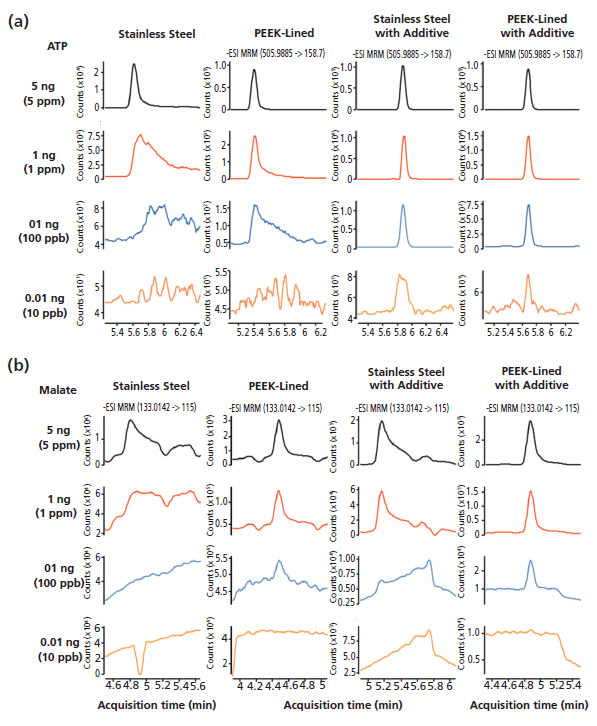
Figure 5: 1-µL injections of (a) ATP or (b) malate at indicated concentrations were analyzed with HILIC-Z particles packed in either stainless steel or PEEK-lined column hardware. The efficacy of the deactivator additive was demonstrated to enhance the chromatographic performance of analytes with multiple phosphate and carboxylate groups.
HILIC LC–MS Analysis of Cell Culture Media
To test our optimized HILIC LC–MS method in a real-world application, an experiment was designed to monitor nutrient consumption and metabolic waste product secretion in cell culture media after six days (Figure 6). As expected, lactate accumulation was observed in the growth media as a metabolic waste product (Figure 6a, column 1). Moreover, other organic acids associated with the TCA cycle (that is, malate, α-ketoglutarate [α-KG], glutamate, and citrate) were excreted and found to accumulate in the cell culture media (Figure 6a, columns 3–6). In contrast, glucose levels decreased over time as the cells consumed the sugar to fuel their cellular metabolism until the glucose was either completely depleted or below the limit of detection (Figure 6a, column 2). Amino acid levels also decreased over time, which correlated with the consumption of the nutrients from the growth media by the cells (Figures 6b and 6c). Interestingly, the cysteine dimer, cystine, was detected in the cell culture media but not monomeric cysteine (Figure 6c, column 7). Further investigation into the formulation of the culture media revealed that cystine is supplemented in the culture media and not the cysteine monomer. These results demonstrated that in a single HILIC LC–MS run, a wide range of metabolites including organic acids and amino acids could be profiled and monitored from mammalian cell culture media.

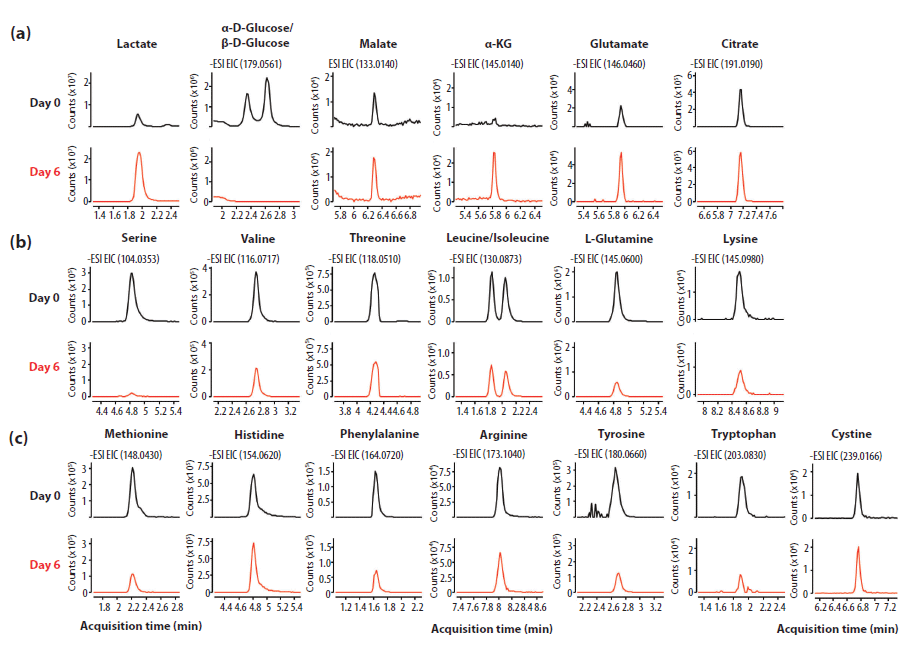
Figure 6: 100 µL of growth media was mixed with 400 µL of 50% acetonitrile. The sample was centrifuged at 10,000g for 5 min and 0.5 µL of the sample was subjected to LC-MS analysis. The sample was analyzed on a 150 mm × 2.1 mm PEEK-lined HILIC-Z column with 10 mM ammonium acetate buffer, pH 9, and 5 µM of the deactivator additive. The connection capillaries were PEEK-lined with a 1290 binary pump coupled to a 6545 Q–TOF system. Runs were repeated in triplicate and the experiment was conducted in negative analysis mode.
Summary
We have developed a HILIC zwitterionic phase superficially porous particle (SPP) that is chemically stable over a pH range of 3–11. The wide operating pH range enables chromatographers to study a broad range of metabolites including underivatized carbohydrates, amino acids, organic acids, polyamines, phosphorylated nucleotides, and sugar phosphates. Importantly, the HILIC chemistry was shown to successfully resolve difficult to separate structural isomers such as leucine and isoleucine and citrate and isocitrate.
For many metabolites commonly studied in the TCA cycle and glycolysis pathway, we found that high-pH mobile-phase buffers greatly improved the signal and peak shape of these metal-sensitive analytes. The use of bioinert hardware helps decrease the amount of contact between metal and targeted metabolites, and thus results in better peak shape and sensitivity. Moreover, we discovered a mobile-phase additive that limited the metal–analyte interaction in the sample flow path and enhanced the chromatographic performance of these metal-sensitive metabolites. This method ultimately facilitated the monitoring of cell culture feedstock (that is, glucose and amino acids) and metabolic waste (such as organic acids) in a complex sample matrix within a single LC–MS analytical run.
References
(1) R.E. Majors, LCGC North Am. 28(4), 8–17 (2010).
(2) V.V. Tolstikov and O. Fiehn, Anal Biochem. 301(2), 298–307 (2002).
(3) A. Masuda and N. Dohmae, Biosci Trends. 5(6), 231–238 (2011).
(4) P. Kiefer, N. Delmotte, and J.A. Vorholt, Anal. Chem. 83(3), 850–855 (2011).
(5) T.O. Metz et al., Biomark. Med. 1(1), 159–185 (2007).
(6) J.W. Dolan, LCGC Eur. 21, 258–263 (2008).
(7) P. Appelblad, T. Jonsson, W. Jiang, and K. Irgum, J. Sep. Sci. 31(9), 1529–1536 (2008).
(8) K. Spagou, H. Tsoukali, N. Raikos, H. Gika, I.D. Wilson, and G. Theodoridis, J. Sep. Sci. 33(6–7), 716–727 (2010).
(9) R. Tuytten et al., J. Chromatogr. A 1104(1–2), 209–221 (2006).
(10) K.T. Myint, T. Uehara, K. Aoshima, and Y. Oda, Anal. Chem. 81(18), 7766–7772 (2009).
(11) J.J. Pesek, M.T. Matyska, and S.M. Fischer, J. Sep. Sci. 34(24), 3509–3516 (2011).
(12) D. Kotoni, I. D'Acquarica, A. Ciogli, C. Villani, D. Capitani, and F. Gasparrini, J. Chromatogr. A 1232, 196–211 (2012).
(13) D.L. Shollenberger and D.S. Bell, LCGC Eur. 29(12), 687–692 (2016).
(14) W.J Long, A.E Mack, X. Wang, and W.E. Barber, LCGC North Am. 33(4), 31–39 (2015).
(15) J.C. Heaton and D.V. McCalley, J. Chromatogr. A 1427, 37–44 (2016).
Jordy J. Hsiao and Genevieve C. Van de Bittner are with Agilent Technologies, Inc., in Santa Clara, California. Andrew P. Kennedy and Ta-Chen Wei are with Agilent Technologies, Inc., in Wilmington, Delaware. Direct correspondence to: jordy.hsiao@agilent.com













Determining the Link Between Prenatal Cannabis Use and Symptoms of Depression Using LC–MS/MS
April 16th 2025Researchers investigating the relationship between cannabis use during pregnancy and depressive symptoms—and whether continued use beyond the first trimester or higher levels of use were linked to increased symptoms—used liquid chromatography–tandem mass spectrometry (LC–MS/MS) to confirm the presence of 11-nor-9-carboxy-delta-9-tetrahydrocannabinol (THC-COOH) in urine samples.



Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

Identifying PFAS in Alligator Plasma with LC–IMS-HRMS
April 15th 2025A combination of liquid chromatography ion mobility spectrometry, and high-resolution mass spectrometry (LC–IMS-HRMS) for non-targeted analysis (NTA) was used to detect and identify per- and polyfluoroalkyl substances (PFAS) in alligator plasma.

