Troubleshooting Basics, Part 3: Retention Problems
What causes peaks to appear where they do not belong?
This is the third instalment in a series focusing on some of the basic principles of troubleshooting liquid chromatography (LC) methods. First, we looked at some general practices for troubleshooting any LC problem (1). Then we looked at problems whose symptoms are related to pressure changes (2). This month, we'll concentrate on problems exhibited as abnormal retention times. As a means to organize the discussion, let's look at situations where retention times are too long, too short or inconsistent.
What Controls Retention?
Before we look at specific problems, let's take a moment to consider the things that influence retention. We can categorize these as the mobile phase, the stationary phase (column), the hardware, the environment and the sample. Let's simplify this discussion and assume that nothing has happened to the sample, such as degradation or other chemical changes.
The mobile phase can change because of an error in formulating it, such as a mistake in volumetric measurement or adjustment of the pH. If an error in formulating the mobile phase is suspected, it is best to make a new batch to see if it fixes the problem. Some mobile phases can change over time because of chemical degradation, selective evaporation of one component or microbial growth in highly aqueous mobile phases. Again, reformulation is the best way to verify this problem source. Most of us use on-line mixing to prepare isocratic mobile phases. An error in instrument settings or hardware failure can be the cause of errors in on-line mixing. Check for proper degassing and pump operation, as well as the correct control-programme settings. Sometimes hand-mixed mobile phases can be used as a check for on-line mixing, or alternate mixing channels can be used for both isocratic and gradient methods (for example, use the C and D solvent reservoirs instead of A and B in a four-solvent LC system).
The stationary phase in the column has a finite lifetime, generally in the 500–2000 sample range (or more), depending on the nature of the sample. Every column will die eventually, and some methods are harder on columns than others. For example, mobile phases outside the pH 2–8 region accelerate the degradation of silica-based columns. Some column types have shorter lifetimes than others. For example, cyano and amino columns are unlikely to last as long as C8 or C18 columns, which tend to be quite robust. In addition to changes in retention, column failure usually is accompanied by a rise in system pressure and an increase in peak tailing. Replacement of a suspect column with a new one is the easiest way to check for column-related problems.
System hardware problems that generate symptoms of changed retention most commonly are associated with pump malfunctions or leaks. Pump problems can be checked with a simple flow-rate measurement with a stop watch and volumetric flask. A secondary symptom of pump problems may be high, low or fluctuating pressure. Low flow may be associated with faulty check valves, worn pump seals, air bubbles in the pump or errors in pump settings. Cleaning, component replacement or degassing should correct such problems. High flow rates usually are a result of improper settings.
The most common environmental cause of retention changes is a change in column temperature. This effect is common if the column oven is not used or is not working properly. Methods that operate under ambient conditions are highly susceptible to failure, especially if the laboratory temperature is not well controlled. In my travels I have encountered laboratory temperatures ranging from 10 °C (central China in January) to 35 °C (Tel Aviv in June). If we use the rule of thumb that retention can change by 2% with each 1 °C change in temperature, you can imagine the result if the same method were run in both of those laboratories under ambient conditions! Use the column oven and make sure that it is operating properly.
Two Important Measurements
One tool that can be very useful in diagnosing the source of retention problems is the retention factor (also called the capacity factor, κ'). Recall that the retention factor, κ, is calculated as


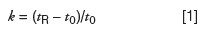
where tR is the retention time and t0 (sometimes abbreviated as tM) is the column dead time, usually measured by the first disturbance in the baseline (the "solvent front"). Another useful calculation is the selectivity, or relative retention, α


where κ1 and κ2 are the κ-values for the first and second peaks of an adjacent peak pair, respectively.
Notice that changes in flow rate, whether intentional or due to a leak, will change both t0 and tR proportionally, so κ will remain constant for such changes. On the other hand, chemical changes will change only tR, so the κ value is changed, too. Generally, when the κ value is changed it does not change exactly the same for all peaks in the chromatogram. One way to confirm chemical changes in the system is to check the α value; if α changes, a chemical source of the problem is most likely. Because κ and α are so useful in distinguishing between flow-related and chemical changes, it is a good idea to make these measurements a part of the process for troubleshooting retention-time problems.
Excessive Retention
When retention times increase and κ-values stay constant, a flow-rate problem is indicated. Double-check the flow-rate setting to be sure you didn't make a mistake. Leaks and pump problems are the two most common remaining causes. Check for leaks throughout the system; these may or may not be accompanied by low system pressure, depending on the magnitude of the leak. Several possible problems related to the pump could be sources of increased retention. Air bubbles in the pump will also cause pressure fluctuations; thorough degassing of the mobile phase and purging the pump should correct such problems. If problems persist, check to be sure the frit in the mobile phase reservoir is not restricting flow to the pump. Faulty check valves or pump seals also can result in low flow and long retention times. Sonication of check valves will usually restore their function. Pump seal leakage often is accompanied by liquid dripping from the drain hole just behind the inlet check valve on most pumps. Check the maintenance records — if the pump seal has been in use for a year or more, replace it.
When a change in κ values (and often α) is observed, you have evidence that a change in system chemistry is responsible for an increase in retention. The easiest way to check this is to make a new batch of mobile phase. If this does not correct the problem, replace the column.
A final possible source of increased retention is a drop in the column temperature. As mentioned above, a 2% increase in retention for a 1 °C drop in temperature is common. Based on the magnitude of the retention change, you can estimate the temperature change and see if it is a reasonable cause of retention. Has the oven failed, did you forget to turn it on or are you relying on ambient operating conditions? Any of these sources can account for increased retention.
Retention Is Too Small
When retention times are smaller than they normally appear, a flow-rate change is highly unlikely, unless you made an error in one of the settings. This is because decreases in flow due to leaks or other malfunctions are not uncommon, but there are no corresponding causes that result in higher-than-normal flow rates that are necessary for reduced retention.
As with retention times that are too long, do a stepwise elimination of problem sources by first making up a new batch of mobile phase. If this approach doesn't fix the problem, replace the column. Temperatures that are higher than normal will cause a drop in retention; the sources of temperature problems are the same as for excess retention.
Fluctuating Retention Times
Usually, an increase or decrease in retention will not be an abrupt change. If it is, the cause is likely related to operator intervention, such as improper formulation of a new batch of mobile phase, installing the wrong column or changing a column-oven setting. More commonly, retention will gradually increase or decrease over tens, hundreds or thousands of samples, or it cycles over a 24-h period. Cycling retention is commonly correlated with ambient methods and a laboratory temperature that changes throughout the day and night.
Retention drift that occurs over hundreds or perhaps thousands of injections is most likely because of normal column aging. Drift from column aging usually will be accompanied by a gradual increase in pressure and an increase in peak tailing. Often, a shift in relative retention also will be observed when α-values are calculated. If the column is suspected, replace it to see if the problem is corrected.
Shorter-term retention drift may be caused by the mobile phase. Although fairly rare, it is possible to selectively evaporate a volatile component of the mobile phase, especially if helium sparging is used for degassing. Retention drift resulting from a pH shift can occur if the buffer is outside its optimal buffering region, generally ±1 pH unit from its pKa. The use of volatile buffers, as is common with LC–mass spectrometry (MS) mobile phases, may shorten the stable lifetime of the mobile phase, so daily reformulation may be a wise practice. Make up a new batch of mobile phase if the mobile phase is suspected, and be sure to adjust the pH before any organic solvent is added.
Problem Prevention
To avoid retention problems, make sure to keep the instrument in good operating condition by servicing it regularly. A routine preventive maintenance session should be done on an annual basis at a minimum, and more often for heavily used LC systems.
Because column temperature changes can have such a profound influence on retention time, it is wise to always use a column oven. Many ovens do not control the temperature well near room temperature; a good practice is to use a minimum operating temperature of 30–35 °C so that good temperature control is ensured. It may take 30 min or longer for the column oven to reach a stable temperature. Be sure to use the solvent preheater that is included with most column ovens. The most common preheater implementation is a piece of capillary tubing that is embedded in the aluminum block of the oven.
Columns will usually last for more than 1000 injections. When this number of samples has been analysed, the cost-per-sample for the column is less than 5% of the overall per-sample cost of analysis. My feeling is that at this point it isn't worth my time to do anything more than flush the column with strong solvent (for example, acetonitrile or methanol); if this doesn't restore the column, replace it. Guard columns or sample preparation will both extend the column life, but they have their associated costs, which may make the economics of their use questionable. A 0.5-µm in-line filter between the autosampler and column will help keep particulate matter from blocking the column inlet frit, but it will not influence retention-related problems. Another good practice for extending column life is to use a single column for each method. When the same column is used for multiple methods, sometimes unimportant sample components from one method will interfere with another method.
Mobile phases have finite lifetimes too. My recommendation is to replace any buffer at least once a week and organic-based mobile-phase components at least monthly. It is a good idea to replace the reservoir with a clean one whenever the mobile phase is replaced so that prior contamination doesn't get transferred to the new mobile phase.
If you pay close attention to patterns in retention changes, correlations with mobile-phase use and column history, you can establish expected replacement cycles for each component of each method. After such patterns are defined, you can put in place preventive-maintenance and component-replacement practices that will help you avoid most retention-related problems. Armed with an understanding of which variable most strongly influences retention in your particular method, you'll be able to more quickly identify and correct problems when they occur.
"LC Troubleshooting" editor, John W. Dolan, is vice president of LC Resources in Walnut Creek, California, USA. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column should go to "LC Troubleshooting", LCGC Europe, 4A Bridgegate Pavilion, Chester Business Park, Wrexham Road, Chester, CH4 9QH, UK or e-mail the editor, Alasdair Matheson, at amatheson@advanstar.com
References
1. J.W. Dolan, LCGC Europe, 24(7), 386–389 (2011).
2. J.W. Dolan, LCGC Europe, 24(9), 484–489 (2011).















University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.


Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

