Tools for Functional Assessment of Biotherapeutics


Biologically derived therapeutics, such as monoclonal antibodies (mAbs), are playing an increasingly important role in revolutionizing modern medicine for treating cancer and many autoimmune disorders. They have significantly improved health care outcomes for patients because of their superior target specificity. Because the function of the drug is impacted via multiple pathways and perturbations at various levels of the protein architecture, a detailed study of functional assessment by using orthogonal tools has become a necessary part of their characterization. In the case of biosimilars, demonstration of biological comparability to the originator is an essential component of the comparability package that the manufacturer submits to the regulatory authorities. In this article, we present many of the various tools that are routinely used for performing such functional characterization of biotherapeutic products. Traditional cell-based approaches include measurement of the antibody-dependant cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP). More recently, approaches based on ligand binding, including fluorescence-activated cell sorting (FACS), surface plasmon resonance (SPR), and biolayer interferometry (BLI) are commonly used as well. Current understanding of the role these approaches can play in a functional assessment has been elucidated.
Therapeutic biologics are becoming a critical and significant component of drug discovery pipelines in the pharmaceutical industry. The utility of biologic drugs for treating unmet medical needs is being expanded beyond oncology and immunology into other therapeutic areas, including cardiovascular and metabolic diseases (1). Drug disposition of biotherapeutics is vastly different from small molecules (2) and therefore, assessment of their safety and efficacy is critical for clinical development. Determining the safety of biotherapeutics often involves evaluating multiple factors, including those related to the product, patient, and treatment (3,4). Another trend is the rise of biosimilars, driven by the societal needs of affordable and accessible biotherapeutics. This has further necessitated the need for a meticulous and rigorous risk–benefit analysis of the biologics, involving establishing comparability of biosimilars. There are instances where process and formulation changes have been introduced to ensure similar quality, safety, and efficacy in line with the expectations, and a slew of guidelines have been issued by the regulatory authorities in this regard (2,5).
Several researchers have reported on development of platform methods or approaches for demonstrating analytical comparability with the reference product. Characterization of therapeutic monoclonal antibodies (mAbs) involves using a wide range of analytical techniques, along with ligand-binding and cell-based potency assays (6). Because publications on analytical comparability are commonplace, those on functional comparability are limited to determining either binding affinity to the receptors or the potency (depending on the mechanism of action) of the biosimilar on the relevant cell lines. Thus, considering the complexity of the therapeutic drug and the significant possibility that the function of the drug might be impacted via multiple pathways at various levels of the protein architecture, assessment of functional comparability is as important as analytical comparability.
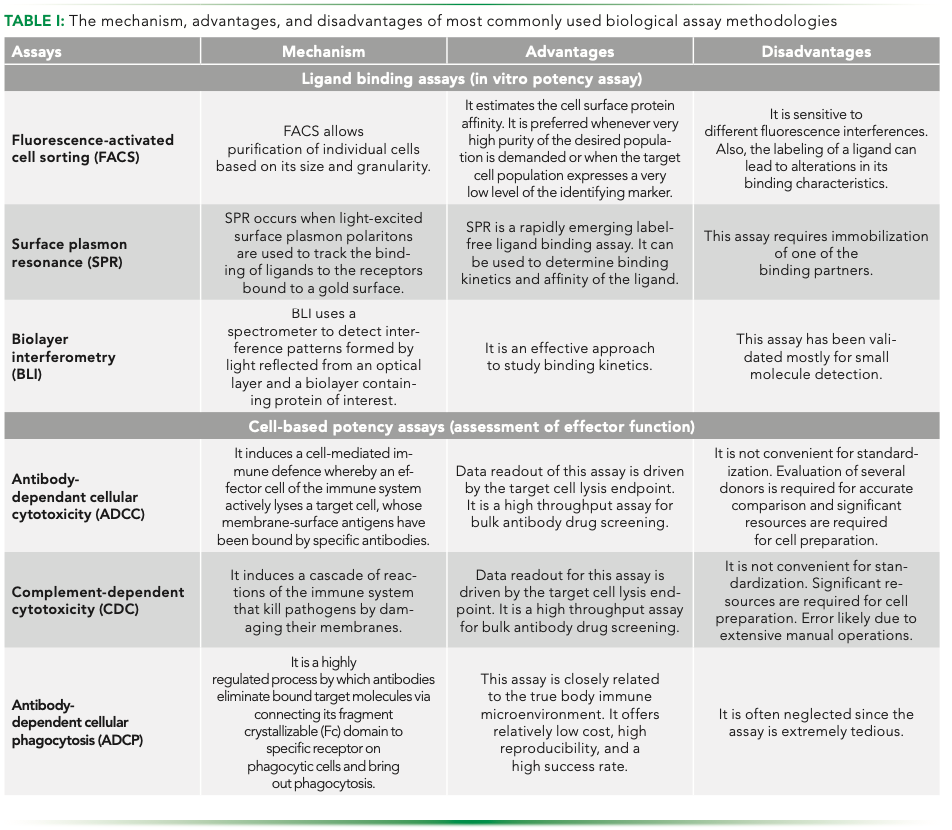
Each functional assay has its pros and cons and hence just like in the case of analytical comparability, a platform of complementary assays is utilized. Table I summarizes the advantages and disadvantages of the most commonly used functional assays.


Design of bioassays for mAbs is driven by a physiological mechanism of action (MOA). Bioassays are always unique for each therapeutic, unlike other analytical techniques, and a well-designed functional assessment can precisely capture the biological activity of the mAb.
Ligand-Binding Assays (In Vitro Potency Assay)
Protein–protein interactions play a major role in the formation of protein complexes, which is the basis of various biological processes. The quantification of ligand binding to specific receptors is the key for drug development research, specifically for determining biological potency. The important aspects of ligand–receptor binding interactions include binding affinity and kinetics, thermodynamics, and ligand efficiency (7). Every step of ligand–target interactions can be studied by multiple binding assays.
Fluorescence-activated cell sorting (FACS) is the adopted method used to obtain a high purity sample of the product of interest. It allows purification of individual cells based on size, granularity, and fluorescence. It gives the percentage, actual number of cells, and mean fluorescence intensity (MFI) of the cell population, and can measure multiple parameters simultaneously on hundreds of individual cells per second; it is a powerful technology with a wide variety of applications in biopharmaceutics (8,9). Appropriate fluorochromes, dyes, conjugates, incubation temperatures, and periods are the essentials for cell surface protein estimation to get well-defined data from the samples.
Two prime tools that have gained prominence for establishment of functional comparability are surface plasmon resonance (SPR) and biolayer interferometry (BLI). Both techniques are optic-based label-free techniques that are used for estimating the biomolecular interaction profiles of biosimilar drugs. These two techniques are superior to traditional fluorescence or luminescence techniques, that are aided by labeling, because labeling is cumbersome and can occupy principal binding sites of the interacting molecule, thereby leading to conformational changes in the molecule and increase non-specificity in the analytical result (10,11).
SPR allows real-time, label-free detection of the interactions between biomolecules. Surface plasmon resonance is a phenomenon that occurs when polarized light strikes an electrically conducting surface at the interface of two media. This interaction generates plasmons, which are electron charge density waves, reducing the intensity of reflected light at a specific angle, referred to as the resonance angle. SPR is a powerful technique for measuring the binding of any pair of interacting molecules, including proteins, antibodies, and antigens. Interactions are measured in real-time, which enable the determination of kinetic parameters. The characterization of antibody–antigen interactions is essential for the successful development of biotherapeutics (9). For about two decades, SPR biosensors have been used to characterize antibody–antigen interactions with applications ranging from the low-resolution affinity screening of antibodies to the high-resolution kinetic constant determinations of the antibodies, and the classification of antibody binding epitopes via epitope-binding studies (12).
BLI works on the principle of superimposition of electromagnetic waves of similar or different phases (13,14). BLI platforms comprise two interfaces, one between the glass fiber and the proprietary biomolecule, and the other between the surface chemistry and the solution. In addition, the BLI platform includes a disposable dip and an optical biosensor. Immobilization of a biomolecule on the surface tip of the glass fiber increases the pathlength of the reflection at the interface between surface chemistry and the solution that changes the interference patterns of all the wavelengths. When the interferometric profiles of the wavelengths are plotted, that results in a new profile that exhibits a shift to the right compared to the original profile. Therefore, if the immobilized molecule interacts with another molecule, there will be a further shift in the interferometric profile. This provides real-time kinetics and quantitation data of biomolecular interactions without the use of labeling (13,14).
Cell-Based Potency Assays
Although the ligand-binding assay can be performed readily with great precision and accuracy, the MOA typically involves post-ligand-binding and thus, the binding activity alone is an incomplete measure of potency. Evaluation of potency at the cellular level is required based on the understanding of the MOA for different targets. The biotherapeutic can typically either induce an early response (signaling pathway) or a late response (proliferation, cytokines).
MAbs target soluble receptors or cytokines on the cell surface and evaluation of this interaction can be achieved from cytotoxic assays including apoptosis to cell proliferation and metabolic assays. The antigen-biding fragment (Fab) fragment of the mAb is mainly associated with binding specificity, while the antibody fragment crystallizable (Fc) portion is for the function of immunoglobulin G (IgG) at the cell level (15). There is significant discrepancy between the potency of mAb therapeutics measured in vitro and in vivo in regards to the required doses. Apart from complement activation, other effector functions are also important for MOA of mAbs.
As molecular-targeted biotherapeutics, antibodies that bind to specific cell-surface antigens on target cells can induce cytotoxicity via the effector functions of antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) through the constant region of the Fc, and apoptosis of target cells (15,16). Establishment of an ADCC or CDC assay should be performed according to the characteristics of the mAbs because the assessment of effector functions is important for the development of the original mAbs candidates. Both in vitro assays are common tools for immunotherapeutic drug discovery and biosimilar development.
ADCC is a lytic mechanism that can be mediated by autoreactive IgG where monoclonal antibodies are able to elicit killing of antibody-coated target cells. The autoreactive IgG recruits effector cells to the target cells with the Fab portion of the IgG binding to the target cell and the Fc region associating with Fc receptors (FcR) on effector cells such as natural killer (NK) cells, monocytes, macrophages, neutrophils, eosinophils, and dendritic cells. Then, the Fc–FcR cross-linking results in the formation of an immune synapse leading to direct tumor cell lysis through the release of cytotoxic molecules (17).
CDC is a cytolytic cascade mediated by a series of complement proteins generously present in the serum. It is triggered by binding of complement component 1q (C1q) to the constant region of cell-bound antibody molecules (17,19). The first step of the complement cascade is binding of the C1q component to the Fc region, which affects the intensity of the following complement activations. A few approaches have succeeded in enhancing the CDC by facilitating the binding of the antibody constant region to the C1q component. As a result of the engineered amino acid mutations that have been inserted into either Fc or the hinge region, an improvement in C1q binding is observed (20).
The ADCP assay is based on using peripheral blood mononuclear cell (PBMC)-derived macrophages as effector cells. Monocytes are isolated from PBMCs and differentiated to macrophages in culture. The workflow is more sophisticated than ADCC and usually takes more than a week to retrieve macrophages. A dose-dependent curve is generated to assess the ADCP potency and phagocytosis is analyzed using flow cytometry. The ADCP assay is a powerful tool for the assessment of biocompatibility as well as early phage confirmation of in vivo (21–23).
Summary and Perspectives
Because of the complexity and intrinsic heterogeneity of mAbs, extensive physicochemical and biological characterization must be carefully conducted. In this article, we present the major tools that are used for functional characterization of mAb therapeutics, assessment methods and technologies that are used to characterize mAb-based candidate during preclinical and clinical studies. An important step in the successful development of a biosimilar is to establish robust analytical and functional comparability with the innovator (24). These are necessary for the biosimilar manufacturer to take advantage of the significant reduction in clinical data required for achieving regulatory approval (25). Functional tools have played an important role and have gradually emerged as a major resource for characterization of biosimilars, thereby playing a prime role in the biosimilar development. A comprehensive set of bioanalytical methods is typically used to analyze functional integrity and comparability (26, 27).
Establishment of cell-based potency assays is more useful than developing a ligand-binding assay because cell-based assays enable better detection of chemical modifications, such as deamidation in the complementarity-determining region or the Fc region of the molecule on its potency (28). Therefore, cell-based assays should be primarily chosen for product characterization, even for lot release, during the clinical development of mAb-based drug and post-licensure life-cycle management (29,30). However, cell-based methods are rather time-consuming and laborious and offer limited automation possibilities. Moreover, the detection format usually requires a label and a complex labelling protocol (9). The development of ligand-binding assays, used orthogonally, may prove to be a powerful and complementary tool in basic research, drug discovery and development, and downstream bioprocessing.
Competing Interests
No potential conflicts of interest were disclosed.
References
(1) A.S. Rathore, Trends in Biotechnology 27, 698–705 (2009).
(2) M. Hassanein, M.A. Partridge, W. Shao, and A. Torri, Bioanalysis 12(18), 1325–1336 (2020).
(3) A.M. Goetze, M.R. Schenauer, and G.C. Flynn, mAbs 2(5), 500–507 (2010).
(4) A. Eon-Duval, H. Broly, and R. Gleixner, Biotechnol. Prog. 28(3), 608–622 (2012).
(5) S.K. Singh, G. Narula, and A.S. Rathore, Electrophoresis 37(17–18), 2338–2346 (2016).
(6) A.S. Rathore, I.S. Krull, and S. Joshi, LCGC North Am. 36(11), 814–822 (2018).
(7) T. Aung, B. Chapuy, D. Vogel, D. Wenzel, M. Oppermann, M. Lahmann, T. Weinhage, K. Menck, T. Hupfeld, R. Koch, L. Trumper, and G.G. Wulf, PNAS 108, 15336–15341 (2011).
(8) I. Jilani, S. O’Brien, T. Manshuri, D.A. Thomas, V.A. Thomazy, M. Imam, S. Naeem, S. Verstovsek, H. Kantarjian, F. Giles, M. Keating, and M. Albitar, Blood 102(10), 3514–3520 (2003).
(9) M. Cedeño-Arias, J. Sanchez-Ramirez, R. Blanco-Santana, and E. Rengifo-Calzado, Sci Pharm. 79(3), 569–581 (2011).
(10) D. Frenzel and D. Willbold, PLoS One 9(9), e106882 (2014).
(11) A.S. Rathore, I. Krull, J. Batra, and D. Kumar, LCGC North Am. 35(12), 870–877 (2017).
(12) A.S. Rathore and R. Dash, BioPharm Int. 32(3), 34–39 (2019).
(13) X.R. Jiang, A. Song, S. Bergelson, T. Arroll, B. Parekh, K. May, S. Chung, R. Strouse, A. Mire-Sluis, and M. Schenerman. Nat Rev Drug Discov. 10, 101–110 (2011).
(14) P. Hariharan, Basics of Interferometry (Academic Press, Massachusetts, 2007), pp. 3–5.
(15) R. Barbour and M.P. Bova, Bioanalysis 4, 619–622 (2012).
(16) X. Wang, Z. An, W. Luo, N. Xia, and Q. Zhao, Protein Cell 9(1), 74–85 (2018).
(17) M. Tada, A Ishii-Watabe, T. Suzuki, and N. Kawasaki, PLoS One 9(4), e95787 (2014).
(18) N. Nupur, N. Chhabra, R. Dash, and A.S. Rathore, mAbs. 10(1), 143–158 (2018).
(19) S. Lida, R. Kuni-Kamochi, K. Mori, H. Misaka, M. Inoue, A. Okazaki, K. Shitara, and M. Satoh, BMC Cancer 9, 58 (2009).
(20) S. Lida, H. Misaka, M. Inoue, M. Shibata, R. Nakano, N. Yamane-Ohnuki, M. Wakitani, K. Yano, K. Shitara, and M. Satoh, Clin. Cancer Res. 12, 2879–2887 (2006).
(21) A.W. Pawluczkowycz, F.J. Beurskens, P.V. Beum, M.A. Lindorfer, J.G.J van de Winkel, P.W.H.I. Parren, and R.P. Taylor, J. Immunol Methods 183(1), 749–758 (2009).
(22) N. Gül and M.V. Egmond, Cancer Res. 75(23), 5008–5013 (2015).
(23) L. Kamen, S. Myneni, C. Langsdorf, E. Kho, B. Ordonia, T. Thakurta, K. Zheng, A. Song, and S. Chung, J. Immunol Methods 468, 55–60 (2019).
(24) A. Ishii-Watabe and T. Kuwabara, Drug Metab Pharmacokinet. 34(1), 64–70 (2019).
(25) E.R. Kabir, S.S. Moreino, and M.K.S. Siam, Biomolecules 9(9), 410 (2019).
(26) A.S. Tsiftsoglou, S. Ruiz, and C.K. Schneider, BioDrugs 27(3), 203–211 (2013).
(27) H. Schellekens, Nat Biotechnol. 22(11), 1357–1359 (2004).
(28) A.S. Rathore, Biomarker Discovery in the Developing World: Dissecting the Pipeline for Meeting the Challenges (Springer, New Delhi, 2016).
(29) T.T. Hansel, H. Kropshofer, T. Singer, J.A. Mitchell, and A.J.T. George, Nat. Rev. Drug Discov. 9, 325–338 (2010).
(30) R. Bansal, R. Dash, and A.S. Rathore, J. Pharm. Sci. 109(9), 2684–2698 (2020).
ABOUT THE CO-AUTHORS

Rozaleen Dash is with the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.


Ritu Jain is with the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.

ABOUT THE COLUMN EDITORS

Jared Auclair is an Associate Dean of Professional Programs and Graduate Affairs at the College of Science at Northeastern University, in Boston, Massachusetts. He is also the Director of Biotechnology and Informatics, as well as the Director of the Biopharmaceutical Analysis Training Laboratory.


Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India. Direct correspondence to: LCGCedit@mmhgroup.com






 Column Gas Chromatography (GC)")















Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

