The Role of Separation Techniques in the Analysis of mRNA Therapeutic Drug Substances and Drug Products
Messenger ribonucleic acids (mRNA) therapeutics are becoming more widespread pharmaceutical tools to treat a wide range of diseases or infections, as highlighted by regulatory approval of two vaccines for SARS‑CoV-2. Alongside their use as vaccines, they also play a role in protein replacement therapy to ensure therapeutic protein is synthesized within the patient. Structural elements, such as the 5’ cap, UTR regions, reading frame, and poly A tail are considered as critical quality attributes (CQAs) that are subject to a range of analytical techniques. However, chromatography and other separation methods are commonly used for characterization and quantification of the drug substance and drug product. This article reviews a range of techniques available for separative analysis of mRNA therapeutics, their associated impurities, and delivery vehicles.
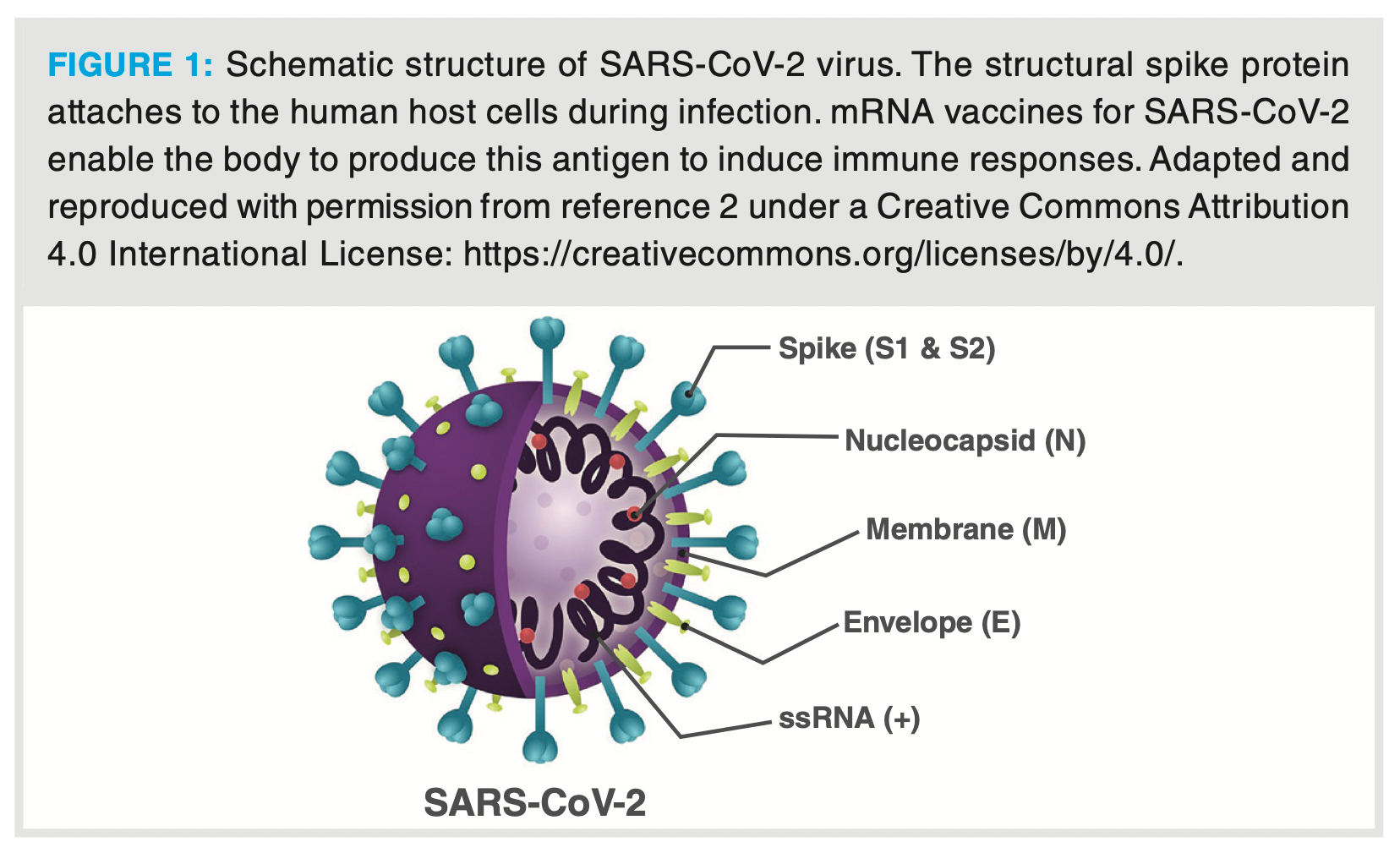
Messenger ribonucleic acids (mRNAs) are large single-stranded polynucleotides present within the cells of the body. mRNA naturally plays a role in the synthesis of proteins by carrying genetic code from the nucleus to the cytoplasm where it is translated into a protein by ribosomes. mRNA can also be used as a modulator of protein production processes to treat diseases in an approach known as RNA therapy. Modulation of protein production can be in the form of producing pathogen-associated antigens that stimulate the immune system (mRNA vaccination) or in the production of proteins that are otherwise lacking due to disease. There are currently two approved mRNA therapeutics that are registered as SARS-CoV-2 vaccines (manufactured by Pfizer/BioNTech and Moderna) (1). These mRNA therapeutics carry the nucleic acid code to produce a protein (spike) antigen from the outer shell of the COVID-19 virus (Figure 1) (2). Once this protein is synthesized, the individuals’ immune system is stimulated to produce an immune response to the antigen, thereby helping with defence against future infection. In addition to their use as vaccines against viruses, other opportunities include the use of mRNA in immune-oncology encoding patient‑specific cancer antigens and inducing immune responses against cancerous cells.


mRNA therapeutics can also be utilized to produce proteins that are endogenously lacking or abnormal due to disease, an approach known as protein replacement therapy. This is a useful alternative to dosing with recombinant proteins, as mRNA-derived proteins are synthesized within the patient’s body. By producing proteins within the patient’s body, all post-translational modifications (PTMs) are host-suitable and will not induce additional PTM-derived toxicity, which can happen with recombinant proteins manufactured in non-human cells (3). In situ production of therapeutic proteins, such as antibodies, can also reduce treatment costs, as the antibodies are produced with the patients’ PTM machinery, which reduces the complexity of release testing of the pharmaceutical (4). In addition, this approach reduces the requirements for rigorous sterile cell culture protein manufacturing processes. Protein replacement therapy remains challenging because of the need for repeated dosing, although there is a promising phase 1 clinical study investigating mRNA protein replacement therapy for cystic fibrosis (NCT03375047).
The greatest opportunity for mRNA therapeutics is the ability to alter their nucleic acid sequence to code for any protein without significant changes to the overall chemistry of the mRNA. This creates a pathway to the treatment of conditions that were otherwise undruggable in the past using small molecules or antibodies. In addition, mRNA therapeutics produce the target protein without causing mutative effects because they do not alter the DNA sequence within the nucleus (5). The greatest challenges currently associated with mRNA therapeutics are (i) immunotoxic effects from introducing foreign nucleic acids into the body and (ii) ensuring the mRNA gets to the target tissue within the body (targeted drug delivery). Despite these challenges, the global mRNA therapeutic market is expected to reach a value of $101 billion by the year 2026 according to recent market reports, with further growth expected (6). A simple search using the keyword “mRNA” on clinicaltrials.gov returns over 2000 clinical studies, which further highlights the growing interest in this therapeutic modality.
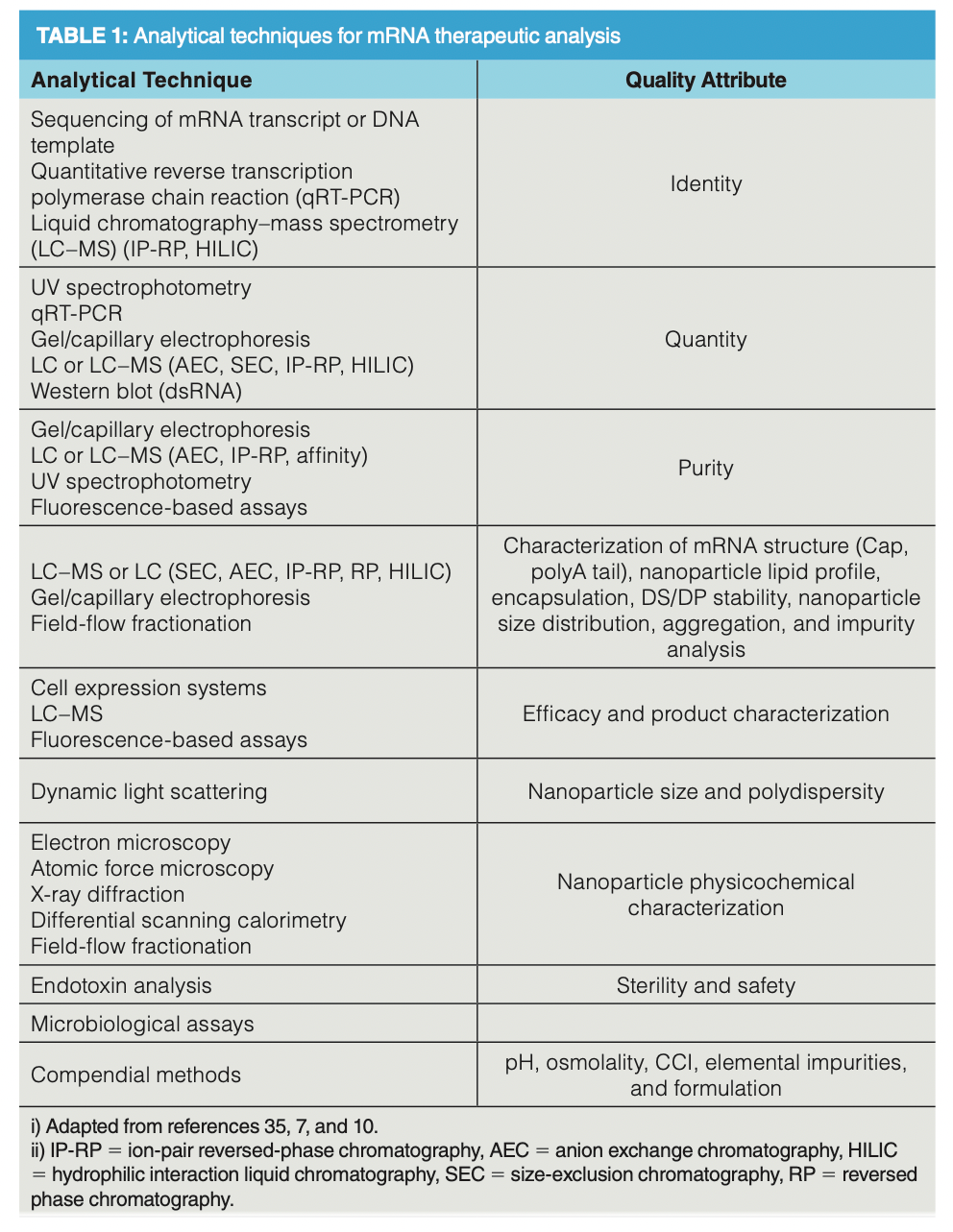
With growing interest comes the requirement for accurate and effective analytical techniques to characterize critical quality attributes (CQAs) of both the drug substance (DS) and drug product (DP). As with other biopharmaceuticals, mRNA therapeutics will be subjected to multiple compendial and non-compendial methods to evaluate identity, appearance, efficacy, toxicology, safety, concentration, pH, osmolality, process-/product‑related impurities, sterility, and formulation (7). Although there is a wide range of techniques available to analyze mRNA CQAs (Table 1), chromatography and other separation techniques sit at the forefront of drug characterization and quantification.

Synthesis
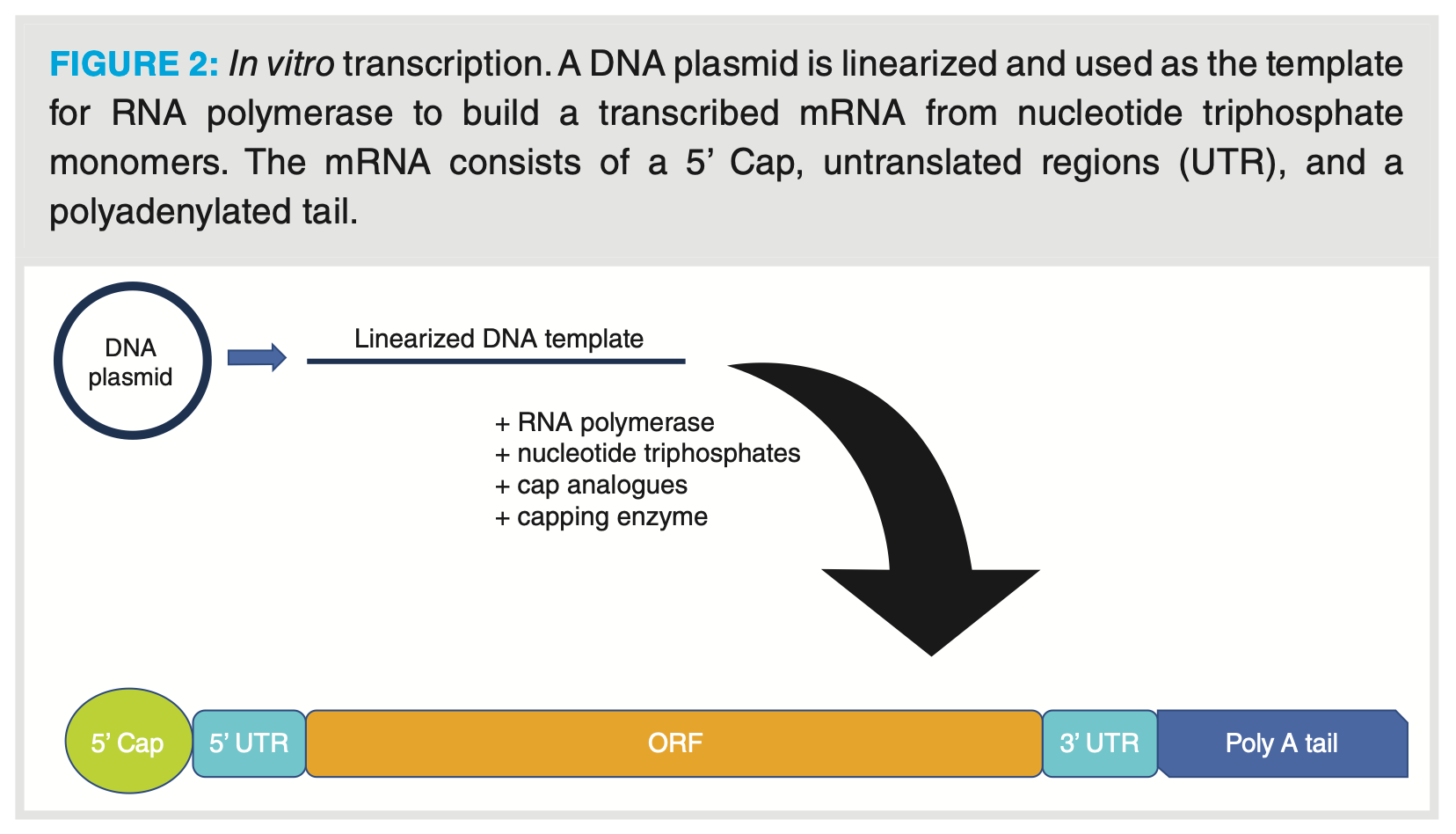
Unlike smaller oligonucleotide therapeutics that are chemically manufactured using solid phase synthesis, mRNA therapeutics are synthesized using in vitro transcription (IVT). This is because solid phase synthesis is limited to producing oligonucleotides smaller than 100 nucleotides, whereas mRNA therapeutics are typically larger than 1000 nucleotides. IVT is performed first by creating a plasmid DNA template that is linearized and transcribed into a mRNA sequence using RNA polymerase and ribonucleoside triphosphates (rNTPs). The DNA template is designed to code for cis-acting structural regions: a 5’ cap structure, 5’ and 3’ untranslated regions (UTR) sandwiching an open reading frame (ORF) containing the protein-encoding sequence of interest, and a poly A tail at the 3’ terminus (Figure 2). The 5’ cap and poly A tail structures assist with splicing, translation mechanisms, and stabilization of the mRNA against degradation, and can be manufactured during the IVT process or enzymatically added post synthesis. mRNA therapeutics are synthesized using a large range of modified nucleosides that help protect against degradation in vivo and reduce the immunotoxicity of the mRNA. Such nucleosides include pseudouridine, N1-methylpseudouridine, 5-methylcytidine, 2-thiouridine, and N6-methyladenosine (4,8).

IVT is challenged by the requirement for a promoter sequence to signal the initiation of transcription by RNA polymerases, which limits the ability to design modifications to the 5’ terminus. In addition, non-specific run-off caused by RNA polymerases can lead to terminal heterogeneity. Despite these challenges, IVT synthesis is robust and reproducible regardless of the sequence of the ORF, thereby allowing rapid alterations to the ORF (and consequently the mRNA product). Rapid alterations to the ORF do not result in drastic changes to the mRNA chemistry, thus giving IVT synthesis a modular, high-throughput capability (1).
Although regulatory guidelines are lacking in regard to mRNA therapeutics, they are classed as advanced therapy medicinal products (ATMPs) by the European Medicines Agency and as biologicals by the World Health Organization (4,9,10). As such, they are subject to quality regulations related to biotechnological products in ICH guidelines, for example, ICH Q5C (stability) or ICH Q6B (specifications) (9). Regulatory considerations for mRNA therapeutics are similar to biologics in the sense of starting material integrity, good manufacturing practice (GMP) manufacture consistency, and quality control of the drug substance/product. Chemistry, manufacturing, and controls management of enzymes, nucleotides, and linear DNA templates used for IVT synthesis is crucial for maintaining quality. CQAs of the DS are mRNA purity, capping efficiency, and heterogeneity of the poly A tail or base sequence. CQAs of the DP are mRNA encapsulation, size distribution and heterogeneity of the delivery nanoparticle, stability, and sterility (11).
Purification
Initial removal of residual double‑stranded DNA template, un-incorporated ribo-nucleoside triphosphates, or enzyme is required to increase DS purity, as their presence can inhibit protein translation, cause immunotoxicity, and affect manufacturing variability and quality (12,13). Although electrophoretic methods can enable purification of the mRNA, the employed denaturing reagents and acrylamide may cause covalent modifications or affect the molecular integrity of the mRNA therapeutic. An alternative approach is the use of size-exclusion chromatography (SEC), which facilitates purification of native state RNA. To remove IVT contaminants, such as the plasmid template, an agarose-based gel matrix was used in conjunction with a mobile phase consisting of 150 mM sodium chloride, 50 mM sodium phosphate, and 0.1 mM EDTA (pH 6.5) (14). Size exclusion columns are also compatible with fast performance liquid chromatography (FPLC) systems for rapid purification (15). However, SEC is limited by low resolution of similarly sized polynucleotides and may not be able to purify all possible fragment contaminants.
Ion-pair (IP) reversed-phase chromatography has also been demonstrated as an efficient approach to remove IVT contaminants (16). mRNAs were purified using a polymeric alkylated, non-porous polystyrene‑divinylbenzene stationary phase and a mobile phase consisting of 0.1 M triethylammonium acetate, pH 7 with 25% v/v acetonitrile as eluting solvent. The effective purification of mRNAs in this way resulted in a 1000‑fold increase in the translation efficiency, indicating the critical nature of the purification step. Other chromatographic approaches may involve affinity-based purification where the mRNA is captured on the solid phase using embedded poly-dT to hybridize with the mRNA poly-A tail (17) or anion exchange (AEC). Chromatographic purification of mRNA is a widely accepted gold standard due to ease of scale-up, and may be coupled with other filtration methods, such as tangential flow filtration or precipitation, to further remove contaminants (12).
Analysis of Drug Substance
mRNA is composed of multiple structural elements that require characterization to determine the DS identity, consistency, and heterogeneity. The 5’ cap structure ensures that the mRNA is recognized and stable against enzymatic degradation. The 5’ cap can be attached to the IVT mRNA transcript during or after synthesis using capping enzymes or by including a dinucleotide cap analogue to the IVT reagent mixture, respectively. As the latter capping approach can lead to mRNA with the analogue in the reverse orientation, the yield can be as low as 50% (18). Although “anti-reverse” cap analogues have been developed to overcome this challenge (19), heterogeneity of the 5’ cap has led to the requirement for accurate 5’ cap characterization. This can be performed by first enzymatically digesting the mRNA and identifying forward and reverse cap structures based upon chromatographic retention time using AEC and radiolabelling the terminal phosphate. To reduce the complexity of capping analysis, non-labelled approaches can be used. One label-free approach utilizes a biotin-tagged RNase H cleavage probe to hybridize with the mRNA 5’ terminus, which was then digested with RNase H. Following sample clean-up, the 5’ cap structures were analyzed using IP reversed-phase liquid chromatography–mass spectrometry (LC–MS); specifically this was demonstrated using a C18 column and a mobile phase consisting of 8.15 mM triethylamine, 200 mM hexafluoroisopropanol (HFIP) (pH 7.9), and methanol as organic solvent (18). Analysis of the cap structures using negative ionization MS facilitated 5’ cap characterization of a range of structures.
Another structural element that can lead to DS heterogeneity is the 3’ poly-A tail, which can be encoded into the template DNA or by enzymatic addition. Poly A fragments can be enzymatically isolated and analyzed using denaturing polyacrylamide gel electrophoretic separation; however, length determination is reliant on accurate comparison to the relative migration of reference ladders within the sample (20). Poly A tail length can also be determined using LC–MS approaches (21,22). RNase T1 was used to cleave the tail and oligo dT magnetic beads were employed to remove the poly A tail fragments from the reaction mixture. The analytes were resolved with IP reversed-phase chromatography on a polymeric reverse‑phase column, using a mobile phase of 15 mM dibutylamine and 25 mM HFIP (with methanol as organic solvent) (22).
The mRNA base sequence is a key quality attribute because it defines the code for protein translation. Errors could lead to erroneous protein production and reduction in therapeutic efficacy. For many years, traditional RNA sequencing approaches were more widely used (23). These approaches were based on the creation of complementary DNA (to the original RNA sequence) libraries and sequencing cDNA fragments using fluorescently labelled nucleotides and next-generation sequencing instruments. However, LC–MS and LC–MS/MS approaches are becoming more common within the bioanalytical laboratory to sequence RNA polynucleotides. The mRNA is enzymatically degraded using RNases and the fragments are identified by their mass using MS. There is a range of available enzymes and often multiple are required to gain higher sequence coverage. One approach has been to use RNase T1, colicin E5, and MazF to generate mRNA fragments for orthogonal mass mapping of mRNAs (24). The challenge with single enzyme digestion is the creation of a large amount of short, isobaric RNA fragments with the same sequence, as these are hard to map to specific points in the RNA sequence. Multiple enzyme digestion overcomes this challenge by creating a wider range of isomeric and unique fragments by digesting at more RNA positions. The mRNA fragments were resolved using 0.1% diisopropylethylamine, 1% HFIP (acetonitrile organic solvent for elution) on an ultrahigh-pressure liquid chromatography (UHPLC) oligonucleotide C18 column. The UHPLC separation enabled sufficient resolution of all of
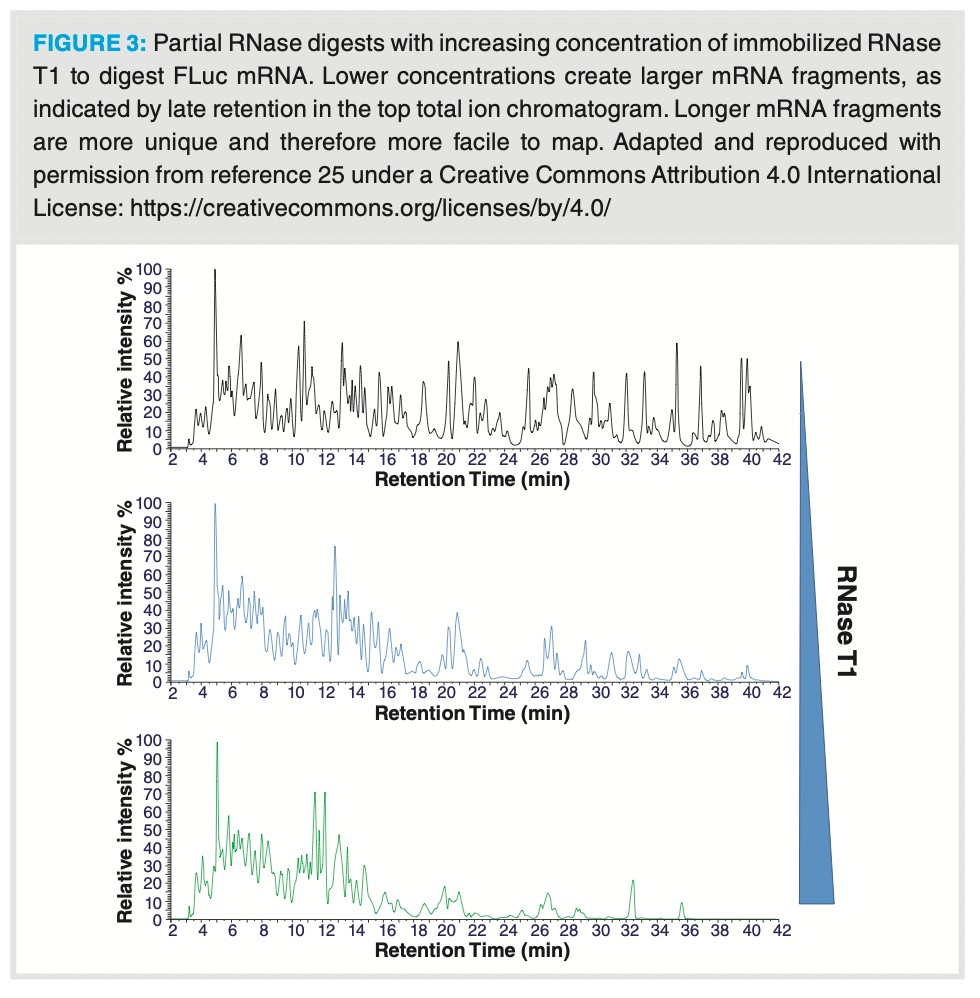
the mRNA fragments and sequential MS/MS sequencing of collisionally activated dissociation fragments. Recent work at the University of Sheffield also applied a mass mapping approach to mRNA sequencing (25). Within this work, mRNA was partially enzymatically digested with RNase T1 to induce missed cleavages and therefore create larger mRNA fragments. The larger, more unique fragments were separated using 0.2% triethylamine, 50 mM HFIP (acetonitrile solvent) on a polymeric reverse-phase column coupled with a high resolution orbital trap mass spectrometer. Figure 3 demonstrates the ability to tune enzymatic digestion with controlled RNase immobilized enzyme on magnetic particles. As fragmentation is not driven to completion, longer RNA fragments remain, which appear at later retention times using IP reversed-phase chromatography. A key tool in the ability to rapidly sequence the mRNA fragments in thedigest is the use of oligonucleotide-appropriated data processing software that has the capacity to sequence modified oligonucleotides from MS data. Another key objective in any approach to sequence mRNA by mass mapping is to optimize the chromatographic separation prior to MS detection to reduce ionization inhibition and increase sensitivity. IP reversed-phase chromatography can be challenging to couple with MS, as the ion-pair reagent decreases sensitivity by suppressing electrospray ionization efficiency (26). Optimization of sensitivity may be achieved by optimizing the mobile phase pH and selecting an appropriate ion-pair reagent, or with the addition of an acidic fluoroalcohol (such as HFIP) to reduce the pH of the mobile phase. These approaches can narrow the oligonucleotide fragment MS charge state distribution and increase oligonucleotide fragment resolution. An alternative chromatographic approach is to employ hydrophilic interaction liquid chromatography (HILIC) to resolve ribonucleotide fragments after enzymatic degradation. This has been demonstrated using a quaternary ammonium‑bonded polymeric column and a mobile phase containing 15 mM ammonium acetate, pH 5.5 (using acetonitrile as organic solvent and water for elution) (27). The benefits of this approach are the absence of an ion-pair reagent that would reduce MS sensitivity and the maintenance of high chromatographic selectivity for isomeric fragments containing structurally similar nucleobases, such as uridine and pseudouridine, that may exhibit low resolution in other chromatographic conditions. Increased polarity in pseudouridine containing RNA fragments induce stronger retention to the HILIC column.

A method to identify the nucleoside stoichiometric composition of an mRNA used capillary zone electrophoresis (CZE) coupled with tandem MS (28). The mRNA was first digested into nucleosides and separated using a bare fused‑silica capillary and 10% acetic acid solution, pH 2.2. This was followed by tandem MS detection to quantify modified and unmodified nucleosides, which can be an orthogonal approach to RNA mass mapping using LC–MS approaches.
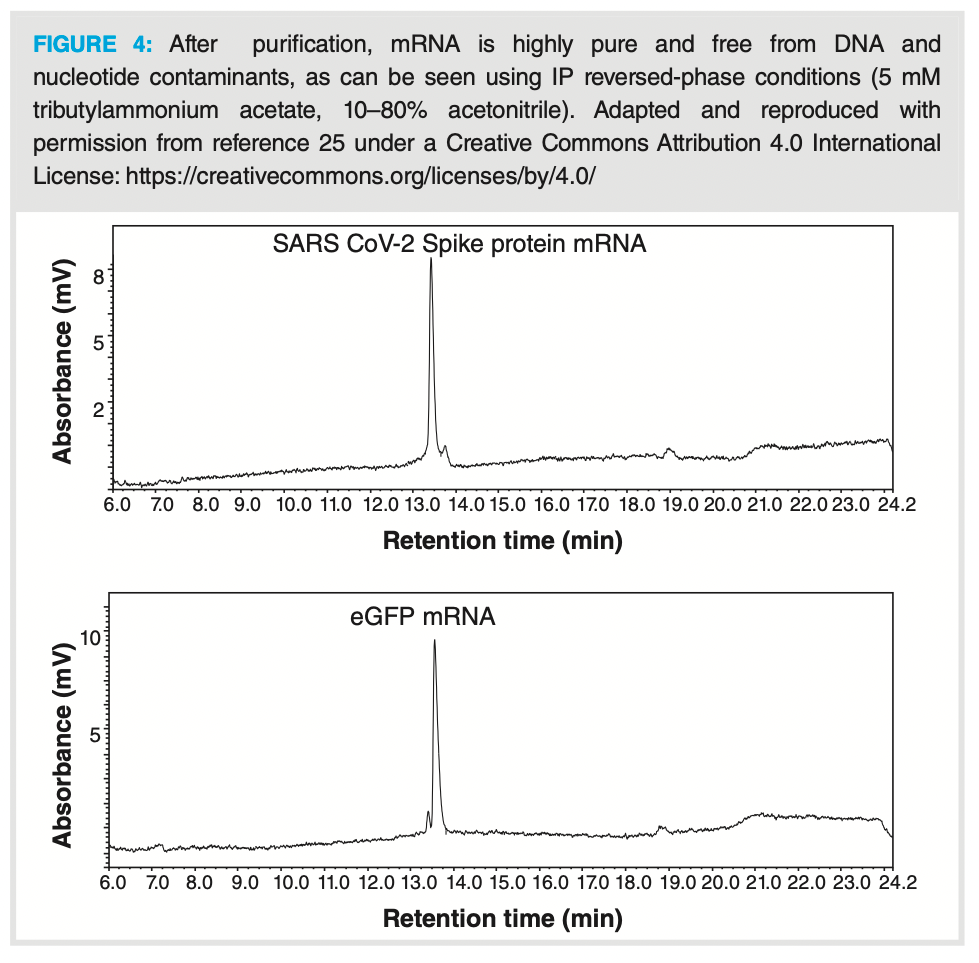
Purity analysis of mRNA is challenging as the longer an oligonucleotide becomes, the harder it is to resolve molecules of similar length chromatographically (29). In addition to IP reversed-phase, anion exchange has been used to determine mRNA purity using a polymeric positively charged column and 10 mM sodium hydroxide to elute mRNA at pH 12 without on‑column degradation (30). Figure 4 displays a chromatogram obtained after mRNA produced by IVT was purified using a silica membrane. It shows the mRNA in a highly pure form—free from small IVT contaminants, such as free ribonucleotide triphosphates. IP reversed-phase and AEC are optimal chromatographic modes to understand RNA stability and purity, as high-temperature IP reversed‑phase chromatography enables highly efficient peaks and RNA is relatively stable in the basic conditions of AEC mobile phases (30,31). Nevertheless, it remains challenging to determine peak purity due to the low ionization efficiency of large nucleic acids and low chromatographic resolution of large RNA structures. Although chromatographic techniques, such as AEC and IP reversed-phase, can identify some synthesis impurities, analyte and impurity adsorption to metal surfaces within the chromatography system can lead to a reduction in chromatographic efficiency and sensitivity (32). Hybrid surface technologies, such as ethylene‑bridged siloxane polymers, that reduce these effects are a welcome addition to improving chromatographic performance.

Capillary gel electrophoresis is an alternative technique that can be utilized to separate large oligonucleotides and understand DS purity. The application of diluted solutions of aqueous hydroxyethylcellulose separation polymer can enable the resolution of large RNA fragments differing by as little as 700 nucleotides (33).
Analysis of Drug Product
mRNA, like all nucleic acids, is negatively charged and therefore incompatible with the lipid membrane bilayer of the cell. To deliver the mRNA therapeutic to its target, this incompatibility must be overcome. This is achieved by encapsulating the mRNA therapeutic into a nanoparticle, which is more amenable to endocytosis or cell delivery. In addition, the lipid nanoparticle protects the mRNA from degradation post dosage. Delivery nanoparticles are composed of cationic lipids or polymers and may also contain peptides for cellular membrane targeting (4). The two registered mRNA vaccines targeting SARS-CoV-2 are formulated within positively charged lipid nanoparticles to enable cell delivery (1), and as of December 2021, 32 clinical trials were registered for lipid nanoparticle-mRNA vaccines against cancer and infectious diseases (34). Within the drug product formulation, there may also be excipients, such as acetic acid, sodium acetate, potassium phosphate, sodium phosphate, or sucrose, for further stability (13,35).
Critical quality attributes for DP formulations include the amount of the encapsulated vs. un-encapsulated mRNA in formulation, composition, or ratio of lipid species within the nanoparticles and the size distribution of the nanoparticles (36,37). Characterization and quantitation of lipid nanoparticle formulations employ capillary electrophoresis (CE) or reversed‑phase chromatography coupled with UV, refractive index, multi-angle light scattering (MALS), evaporative light scattering (ELS), and charged aerosol detectors (CAD). MALS detectors measure the light that is scattered by a sample passing through the detector whilst in solution, whilst ELS detectors nebulize the column efflux into aerosol droplets. These droplets are dried, leaving the dried analyte to pass a detector that measures the amount of scattered light. Charged aerosol detectors are similar to ELS detectors; however, they charge the dried particles and detect them using an ion trap. These types of detector are useful for the analysis of molecules that do not absorb light, such as lipids; therefore, the benefit of using CAD lies in its ability to characterize both lipid and nucleic acid peaks within the same separation and over a larger DP concentration range (13). There is also future potential to use supercritical fluid chromatography (SFC) for lipid analysis, as has been demonstrated for lipidomic studies derived from biological tissues (38). Identification of lipid species of mRNA-loaded nanoparticles using LC–CAD has been demonstrated using a C18 column and reverse phase chromatography, which facilitated the characterization and quantification of the ratio of lipids present within the nanoparticle (39). SEC-MALS and field‑flow fractionation (FFF) have both been used to characterize loaded nanoparticle size heterogeneity (37). FFF enables the separation of analytes without a stationary phase, using a perpendicular field (for example, electric or thermal) to the mobile phase flow, fractionating the solution mixture according to differing analyte mobilities under the field. In addition, FFF-MALS is able to determine nanoparticle morphology, concentration, stability, and aggregation, which are also CQAs of interest (40).
Drug encapsulation analysis often begins with the separation of un-encapsulated mRNA from the nanoparticles and then comparative quantification of the free mRNA by IP reversed-phase or AEC chromatography vs. the encapsulated fraction. IP reversed‑phase chromatography has been shown to resolve lipid nanoparticles from smaller ribonucleotides using a C18-phenyl column and dibutylammonium acetate as the IP reversed-phase ion-pairing agent (41). This approach could also be utilized for lipid nanoparticle-mRNA formulations and may also provide an indication of stability. Stability-indicating methods are crucial for DP characterization to ensure medicine quality over long periods of time. IP reversed-phase chromatography has been employed to analyze the stability of mRNA up to 1000 nucleotides in length using 0.1 M triethylammonium acetate within the mobile phase (and acetonitrile as organic solvent) and a polymeric reverse‑phase stationary phase (31). Under a range of stress-inducing conditions, degradation products were quantified and relative stability was evaluated.
Conclusions
The diversity of the analytical methods used to evaluate mRNA CQAs shows that chromatography plays a leading role in the purification and characterization of mRNA therapeutics. It is poignant to consider that mRNA therapeutic technologies are continually being advanced and developed. With future advancements in the structure and chemistry of mRNA therapeutics, we will continue to require separation techniques to resolve analytes and critical impurities. In addition, effective delivery of mRNA therapeutics to target tissues remains a challenge, which will also lead to further technological advancement of delivery vehicles, thereby adding to the complexity in developing accurate analytical methods. What is exciting is the range of detection methods available for hyphenation in separations of complex mixtures, such as formulated drug product. Chromatographic stationary phases are also being developed to address more complex and challenging separations. With regard to mRNA and other large nucleic acids, it is clear that low resolution of similarly sized oligonucleotides needs to be addressed—possibly by using novel stationary phases and chromatographic conditions. The wide interest in developing analytical methods for mRNA therapeutics is demonstrable from the wealth of literature, and chromatography will continue to remain crucial for purification and analysis of DS and DP.
References
- Webb, C.; Ip, S.; Bathula, N. V.; et al. Current Status and Future Perspectives on MRNA Drug Manufacturing. Mol. Pharm. 2022, 19 (4), 1047–1058. DOI: 10.1021/acs.molpharmaceut.2c00010
- Santos, I.A.; Grosche, V. R.; Bergamini, F. R. G.; et al. Antivirals Against Coronaviruses: Candidate Drugs for SARS-CoV-2 Treatment? Front. Microbiol. 2020, 11, 1818. DOI: 10.3389/fmicb.2020.01818
- Huang, L.; Zhang, L.; Li, W.; et al. Advances in Development of mRNA-Based Therapeutics. Curr. Top Microbiol. Immunol. 2020, Epub ahead of print. DOI: 10.1007/82_2020_222
- Van Hoecke, L.; Roose, K. How mRNA Therapeutics are Entering the Monoclonal Antibody Field. J. Transl. Med. 2019, 17 (1), 54. DOI: 10.1186/s12967-019-1804-8
- Kim, Y. K. RNA Therapy: Rich History, Various Applications and Unlimited Future Prospects. Exp. Mol. Med. 2022, 54 (4), 455–465. DOI: 10.1038/s12276-022-00757-5
- Thakur, R. mRNA: Therapeutics and Global Markets. BCC Research: Online 2021.
- Poveda, C.; Biter, A. B.; Bottazzi, M. E.; Strych, U. Establishing Preferred Product Characterization for the Evaluation of RNA Vaccine Antigens. Vaccines 2019, 7 (4) 131. DOI: 10.3390/vaccines7040131
- Sergeeva, O. V.; Koteliansky, V. E.; Zatsepin, T. S. mRNA-Based Therapeutics - Advances and Perspectives. Biochemistry (Mosc). 2016, 81 (7), 709–22. DOI: 10.1134/S0006297916070075
- European Medicines Agency, Guideline on the quality, non clinical and clinical aspects of gene therapy medicinal products. 2018.
- World Health Organization, Evaluation of the quality, safety and efficacy of messenger RNA vaccines for the prevention of infectious diseases: regulatory considerations. 2021.
- Knezevic, I.; Liu, M. A.; Peden, K.; Zhou, T.; Kang, H. -N. Development of mRNA Vaccines: Scientific and Regulatory Issues. Vaccines (Basel) 2021, 9 (2), 81. DOI: 10.3390/vaccines9020081
- Pardi, N.; Hogan, M. J.; Porter, F. W.; Weissman, D. mRNA Vaccines — A New Era in Vaccinology. Nature Reviews Drug Discovery 2018, 17 (4), 261–279. DOI: 10.1038/nrd.2017.243
- Ouranidis, A.; Vavilis, T.; Mandala, E.; et al. mRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles. Biomedicines 2022, 10 (1), PMC8773365. DOI: 10.3390/biomedicines10010050
- Lukavsky, P. J.; Puglisi, J. D. Large-Scale Preparation and Purification of Polyacrylamide-Free RNA Pligonucleotides. RNA 2004, 10 (5), 889–93. DOI: 10.1261/rna.5264804
- Kim, I.; Mckenna, S. A.; Puglisi, E. V.; Puglisi, J. D. Rapid Purification of RNAs Using Fast Performance Liquid Chromatography (FPLC). RNA 2007, 13 (2), 289–94. DOI: 10.1261/rna.342607
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weisman, D. Generating the Optimal mRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding mRNA. Nucleic Acids Res. 2011, 39 (21), e142. DOI: 10.1093/nar/gkr695
- BIA Separations, Application note AN062: Purification of Messenger RNA by Affinity Chromatography on CIMmultus Oligo dT Column. 2019.
- Beverly, M.; Dell, A.; Parmar, P.; Houghton, L. Label-Free Analysis of mRNA Capping Efficiency Using RNase H Probes and LC-MS. Anal. Bioanal. Chem. 2016, 408 (18), 5021–30. DOI: 10.1007/s00216-016-9605-x
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R. E. Synthesis and Properties of mRNAs Containing the Novel “Anti-Reverse” Cap Analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl(3’-deoxy)GpppG. RNA 2001, 7, 1486–1495.
- Beilharz, T. H.; Preiss, T. Widespread Use of Poly(A) Tail Length Control to Accentuate Expression of the Yeast Transcriptome. RNA 2007, 13 (7), 982–97. DOI: 10.1261/rna.569407
- Beverly, M.; Hagen, C.; Slack, O. Poly A Tail Length Analysis of In Vitro Transcribed mRNA by LC-MS. Anal. Bioanal. Chem. 2018, 410 (6), 1667–1677. DOI: 10.1007/s00216-017-0840-6
- Liau, B. Agilent application note, Analysis of mRNA Poly-A Sequence Variants by High-Resolution LC/MS. 2021.
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20 (11), 631–656. DOI: 10.1038/s41576-019-0150-2
- Jiang, T.; Yu, N.; Kim, J.; et al. Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91 (13), 8500–8506. DOI: 10.1021/acs.analchem.9b01664
- Vanhinsbergh, C. J.; Criscuolo, A.; Sutton, J. N.; et al. Characterization and Sequence Mapping of Large RNA and mRNA Therapeutics Using Mass Spectrometry. Anal. Chem. 2022, 94 (20), 7339–7349. DOI: 10.1021/acs.analchem.2c00765
- Sutton, J. M.; Guimaraes, G. J.; Annavarapu, V.; van Dongen, W. D.; Bartlett, M. G. Current State of Oligonucleotide Characterization Using Liquid Chromatography-Mass Spectrometry: Insight into Critical Issues. J. Am. Soc. Mass Spectrom. 2020, 31 (9), 1775–1782. DOI: 10.1021/jasms.0c00179
- Lobue, P. A.; Jora, J.; Addepalli, B.; Limbach, P. A. Oligonucleotide Analysis by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry in the Absence of Ion-Pair Reagents. J. Chromatogr. A 2019, 1595, 39–48. DOI: 10.1016/j.chroma.2019.02.016
- Yu, Y.; Zhu, S. -H.; Yuan, F.; et al. Ultrasensitive and Simultaneous Determination of RNA Modified Nucleotides by Sheathless Interfaced Capillary Electrophoresis-Tandem Mass Spectrometry. Chem. Commun. (Camb). 2019, 55 (53), 7595–7598. DOI: 10.1039/C9CC03195B
- Gilar, M.; Fountain, K. J.; Budman, Y.; et al. Ion-Pair Reversed-Phase High-Performance Liquid Chromatography Analysis of Oligonucleotides: Retention Prediction. Journal of Chromatography A 2002, 958 (1–2), 167–82. DOI: 10.1016/s0021-9673(02)00306-0
- Kanavarioti, A. HPLC Methods for Purity Rvaluation of Man-Made Single-Stranded RNAs. Sci. Rep. 2019, 9 (1), 1019. DOI:10.1038/s41598-018-37642-z
- Currie, J.; Dahlberg, J. R.; Eriksson, J.; et al. Stability Indicating Ion-Pair Reversed-Phase Liquid Chromatography Method for Modified mRNA. ChemRxiv Cambridge: Cambridge Open Engage. 2021.
- DeLano, M.; Walter, T. H.; Lauber, M. A.; et al. Using Hybrid Organic-Inorganic Surface Technology to Mitigate Analyte Interactions with Metal Surfaces in UHPLC. Anal. Chem. 2021, 93 (14), 5773–5781. DOI: 10.1021/acs.analchem.0c05203
- Todorov, T.I.; de Carmejane, O.; Walter, N. G.; Morris, M. D. Capillary Electrophoresis of RNA in Dilute and Semidilute Polymer Solutions. Electrophoresis 2001, 22 (12), 2442–2447. DOI: 10.1002/1522-2683(200107)22:12<2442::AID-ELPS2442>3.0.CO;2-9
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for mRNA Delivery. Nat. Rev. Mater. 2021, 6 (12), 1078–1094. DOI: 10.1038/s41578-021-00358-0
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J. A.; et al. mRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. DOI: 10.1016/j.ijpharm.2021.120586
- Fan, Y.; Marioli, M.; Zhang, K. Analytical Characterization of Liposomes and Other Lipid Nanoparticles for Drug Delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. DOI: 10.1016/j.jpba.2020.113642
- Zhang, J.; Haas, R. M.; Leone, A. M. Polydispersity Characterization of Lipid Nanoparticles for siRNA Delivery Using Multiple Detection Size-Exclusion Chromatography. Anal. Chem. 2012, 84 (14), 6088–96. DOI: 10.1021/ac3007768
- Miroslav, L.; Holčapek, M. High-Throughput and Comprehensive Lipidomic Analysis Using Ultrahigh-Performance Supercritical Fluid Chromatography–Mass Spectrometry. Analytical Chemistry 2015, 87 (14), 7187–7195. DOI: 10.1021/acs.analchem.5b01054
- Kinsey, C.; Lu, T.; Deiss, A.; et al. Determination of Lipid Content and Stability in Lipid Nanoparticles Using Ultra High-Performance Liquid Chromatography in Combination with a Corona Charged Aerosol Detector. Electrophoresis 2021, 43 (9–10), 1091–1100. DOI: 10.1002/elps.202100244
- Caputo, F.; Mehn, D.; Clogston, J. D.; et al. Asymmetric-Flow Field-Flow Fractionation for Measuring Particle Size, Drug Loading and (In)stability of Nanopharmaceuticals. The Joint View of European Union Nanomedicine Characterization Laboratory and National Cancer Institute - Nanotechnology Characterization Laboratory. Journal of Chromatography A 2021, 1635, 461767. DOI: 10.1016/j.chroma.2020.461767
- Li, L.; Foley, J. P.; Helmy, R. Simultaneous Separation of Small Interfering RNA and Lipids Using Ion-Pair Reversed-Phase Liquid Chromatography. Journal of Chromatography A 2019, 1601, 145–154. DOI: 10.1016/j.chroma.2019.04.061
Christina Jayne Vanhinsbergh is a senior scientist working within the pharmaceutical industry with a focus on therapeutic oligonucleotides and other new modality drugs. Her background is in the analysis of biomolecules, such as proteins and nucleic acids, and her PhD research centred on the development of multidimensional chromatographic methods interfaced with MS detection. Christina was a postdoctoral researcher at The University of Sheffield, developing LC–MS-based analytical methods for the analysis of alkylated nucleic acids and mRNA therapeutics. She has collaborated with multiple industrial partners, and advocates for networking and development opportunities for early career scientists as a committee member of the Chromatographic Society, UK.







In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




