The Role of Adsorption and pH of the Mobile Phase on the Chromatographic Behavior of a Therapeutic Peptide
The impact of two different stationary phases and ion-pair reagents on the retention behavior of glucagon, a therapeutic peptide consisting of 29 amino acidic residues, has been investigated under reversed-phase elution conditions. Retention of glucagon was investigated under isocratic conditions by varying the fraction of the organic modifier in the range of 28–38% (v/v). The two stationary phases have been characterized in terms of excess adsorption isotherms to understand the preferential adsorption of eluent components on them. Results suggest that the ligand characteristics and the pH of the mobile phase play a pivotal role on retention.
Over the last several decades, the use of peptides in the pharmaceutical, nutraceutical, and cosmetic fields has increased substantially. The biopharmaceutical importance of these molecules is that they can selectively interact with a specific receptor, making them potential candidates for use in antitumoral, anticoagulant, antihypertensive, and antioxidant production (1,2).
Therapeutic peptides are mainly produced by liquid- or solid-phase synthesis (3,4). Usually, these production methods lead to a wide range of impurities, and the issue is that the chemical structure of these impurities can be similar to that of the target peptide. Therefore, further processing and purifications are needed to reach purity specifications for pharmaceutical purposes (5). Preparative reversed-phase liquid chromatography (pRPLC) is the most widely used technique for the purification and isolation of therapeutic peptides (6–10). Operative experimental parameters of the purification processes are obtained through trial-and-error strategies, which waste time and product while resulting in operating far from optimal conditions.
When dealing with complex mixtures, a detailed understanding of the fundamentals of the separation process is extremely important to overcome these difficulties. It is well known that the retention behavior of a peptide could significantly differ depending on its concentration, mobile phase composition, ion-pair reagent, and competition for adsorption because of the presence of other molecules (1,2).
In this respect, the investigation of retention mechanisms and thermodynamic equilibrium of the target peptide becomes a crucial and essential tool for the correct design of the separation process because of its clear advantages in regards to time, cost, and ecological impact.
In this study, the effect of the mobile phase composition and the type of ion-pair reagent on the retention mechanism of a therapeutic peptide, glucagon, was investigated and compared on two RPLC columns packed with C18 and phenyl-hexyl fully porous particles (FPPs), respectively. Both columns were designed to handle high-efficiency liquid chromatography (LC).
Materials and Experimental Conditions
All solvents were purchased from Sigma–Aldrich. A 100 × 3.0 mm Supelco Titan C18 column (1.9 μm particle size, 300 m2/g surface area) and a 100 × 4.6 mm Phenomenex Luna phenyl-hexyl (3.0 μm particle size, 400 m2/g surface area) column were used. Uracil (Sigma–Aldrich) was injected for determining the void volume of the columns. Glucagon was obtained from Fresenius Kabi iPSUM.
All the measurements were carried out on an Agilent 1290 Infinity LC system, equipped with a binary solvent pump (max pressure: 1200 bar), a column thermostat, an autosampler, a photodiode array detector (DAD), and a refractive index detector (RID). The RID was used for excess adsorption isotherms determination. The detection wavelength was 220 nm, and the column temperature was set to 25 °C.
Two different mobile phase compositions were used: MP-1 was comprised of water and 0.02% trifluoroacetic acid (TFA) and acetonitrile, whereas MP-2 comprised of water and 20 mM ammonium acetate and acetonitrile.
The retention dependence of glucagon on the amount of organic modifier of MP-1 and MP-2 was performed under isocratic elution conditions in the range between 28 and 38% (v/v) with 1% increments of acetonitrile for both columns. The injection volume was set at 0.5 μL, and the flow rate was set at 0.4 mL/min.
Excess adsorption isotherms have been calculated with a mobile phase made of acetonitrile and water. Temperature was set at 25 °C. The columns were firstly equilibrated with solutions of known composition of acetonitrile in the bulk mobile phase ranging from 0 to 100%. Then, 1 μL of a solution with a slightly different composition with respect to the bulk MP (±1% acetonitrile) was injected. Retention volumes of the perturbation peaks were corrected for the extra-column contribution.
Results and Discussion
Excess Adsorption
Excess isotherms allow one to study the preferential adsorption of the components of the mobile phase (in this case acetonitrile and water) on the surface of the stationary phase. This adsorption leads to changes in the composition of the stationary phase with respect to the bulk mobile phase that profoundly influences retention of analytes (12). Moreover, the study of excess isotherms allows one to describe polar (free silanols) and hydrophobic (coverage density) properties of stationary phases (11–16).
In this study, excess isotherms were calculated by means of the minor disturbance method (12,13). The excess of acetonitrile over water [Γ(c)] was determined from linear perturbations on a series of equilibrium concentrations, when a steady-state equilibrium between mobile and stationary phase has been reached, through the retention volume of perturbation peaks, as follows:


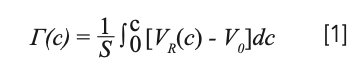
with S the total surface area of the adsorbent in the column (data obtained from manufacturer), VR(c) the retention volume of perturbation peak when the column is equilibrated with a mobile phase containing a concentration c of acetonitrile and V0 the thermodynamic void volume (14). The results are shown in Figure 1 for the two columns.

FIGURE 1: Excess adsorption isotherms and tangent lines on the linear regions for the two columns employed in this work (C18: red, phenyl-hexyl: blue) expressed as μmol/m2 of acetonitrile adsorbed on the stationary phase (ΓexcACN) as a function of the fraction of the organic modifier (ΦACN) in the bulk mobile phase. ACN = acetonitrile.
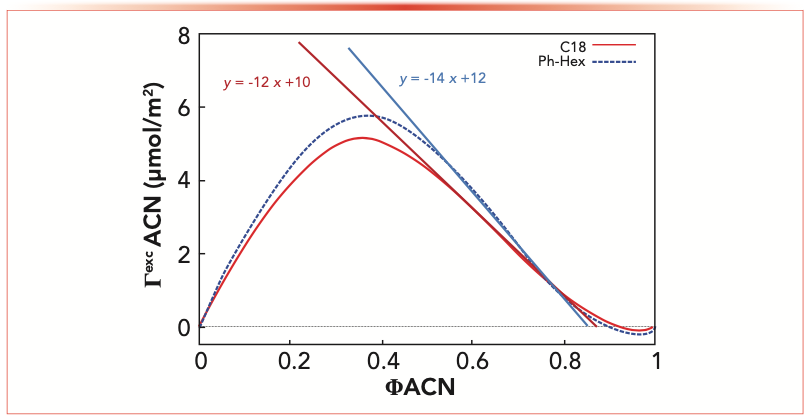
These profiles show an increase of the excess amount adsorbed of acetonitrile on the surface of the stationary phase up to approximately 40% (v/v) of acetonitrile (φACN = 0.4) in the bulk mobile phase, where the maximum value of the excess isotherm is observed. Further increases of acetonitrile bulk concentration lead to a gradual and linear decrease of the excess amount in the region between 50–90% (v/v) (φACN = 0.5–0.9). This behavior occurs because of the saturation of all nonpolar sites present on the surface of the stationary phase with acetonitrile, precluding additional adsorbate accumulation (14). The negative part of the excess isotherms observed at high acetonitrile percentages reveals the preferential adsorption of the second component of the binary mixture, which in this case is water. The amplitude of this region is directly connected to the amount of free silanols present in the surface of the adsorbent accessible by the analyte (11,15,16).
The slope of the inflection tangent line drawn in the decreasing branch of the excess isotherm represents the total amount of the adsorbed phase (Va = b) and the amount of acetonitrile adsorbed (VaACN = a) can be derived from the y-intercept. The tangent line is given by (16) (see Figure 1):

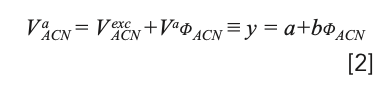
with φACN the volume fraction of the organic modifier in the mobile phase.
It is worth pointing out that the range of validity of equation 2 corresponds to the linear decreasing region of excess isotherm (that is, when adsorption capacity reaches its maximum value) (14).
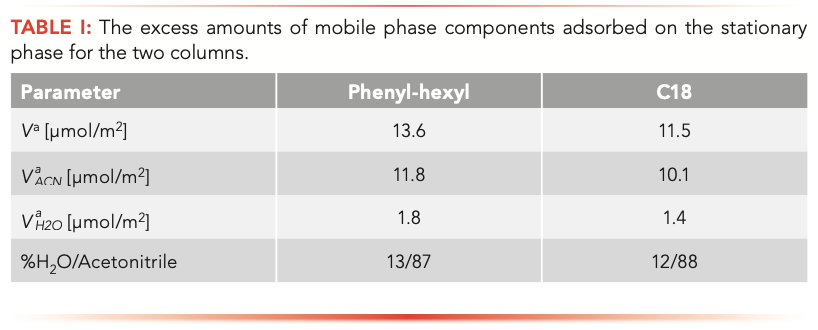
Table I reports data related to excess amounts of acetonitrile and water adsorbed on the stationary phase measured on the two RPLC columns by means of equation 2. From the data, it can be evinced that the total amount of mobile phase, normalized per column surface area, adsorbed on the phenyl-hexyl column is 20% larger compared to the C18 column. Moreover, the phenyl-hexyl column shows a higher amount of adsorbed water (+25%), suggesting a larger presence of residual silanols on the particle surface.

Dependence of Retention of Glucagon on MP Composition
The retention behavior of glucagon at infinite dilution has been studied for MP-1 and MP-2 in the range 28–38% (v/v) of acetonitrile on the two columns. The results are reported in Figure 2 as retention factors of glucagon (k = (tR − t0)/t0, with tR the retention time and t0 the dead time) as a function of the fraction of acetonitrile in the mobile phase. These data demonstrate that, as expected, under the same MP composition, the C18 column shows larger retention when compared to the phenyl-hexyl column, because of its higher hydrophobicity.

FIGURE 2: Dependence of retention factor (k) on the fraction of organic modifier (φ) and pair reagent. For the graph, trifluoroacetic acid (TFA) is green, ammonium acetate is purple; these are measured for C18 (full points) and phenyl-hexyl (empty points) type columns. Note that φACN is the volume fraction of the organic modifier in the mobile phase. ACN = acetonitrile.
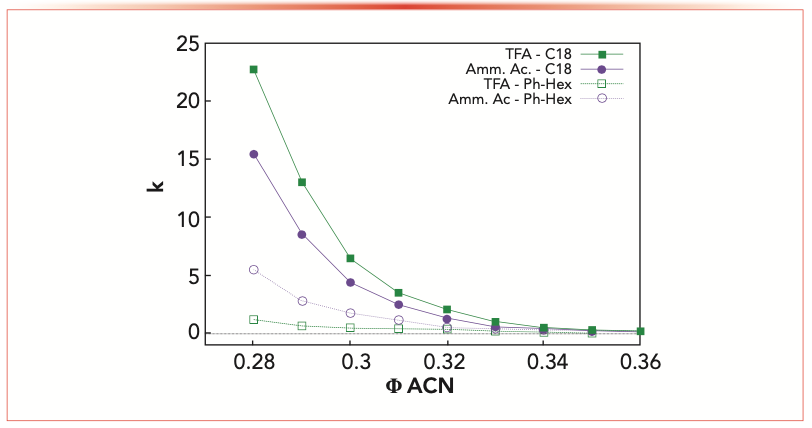
It is well known that retention on the C18 column is to the greatest extent because of hydrophobic interactions between stationary phase and analyte. In addition to hydrophobic interactions, phenyl-hexyl columns are characterized by π-π interactions, because of the presence of phenyl groups. The nature of the mobile phase and the extent of its adsorption on the stationary phase has great impact on retention (by precluding the analyte from establishing interactions with the functional groups of the stationary phase). In this regard, it is plausible that acetonitrile as organic modifier makes the phenyl-hexyl column less retentive for glucagon compared to the C18 column because of the larger presence of acetonitrile on the stationary phase (see Figure 1 and Table I) and π-π interactions between adsorbed acetonitrile molecules and phenyl groups in the stationary phase (17). Surprisingly, a different retention behavior has been observed on the two columns depending on the ion-pair reagent used, as shown in Figure 2. On the one hand, MP-1 containing trifluoroacetic acid (TFA) has led to larger retention on the C18 column (full green squares) if compared to MP-2, which contains ammonium acetate (full purple points), because of the higher hydrophobicity of the complex TFA-peptide. On the other hand, on the phenyl-hexyl column, the most retentive mobile phase was MP-2 (empty purple points) compared to MP-1 (empty green squares).
The opposite retention behavior observed on the phenyl-hexyl column could be because of a combination of the greater accessibility of residual silanols by the analyte and their amount compared to C18 column, the pH of the mobile phase and the strength of the ion-pair reagent.
From excess adsorption results (Table I) it has been evinced that the amount of free silanols on the phenyl-hexyl column is 25% larger with respect to C18 column. Moreover, residual silanols are more analyte accessible on the phenyl-hexyl column, thanks to the rigidity of the ligand itself. Conversely, C18 ligands, having a large degree of freedom, limit the analyte-silanols interactions. These aspects indicate that the effect of free silanols on retention can be more pronounced on the phenyl-hexyl column, while it can be considered negligible on the C18 column.
At a low pH (MP-1), ionization of free silanol groups is sup- pressed and glucagon is positively charged (being its theoretical pIace is between 7.5 and 8.5 (18)). Conversely, when a buffer solution (pH = 7) is used (MP-2), the net charge of glucagon is slightly positive and silanols are deprotonated. In this last case, favorable charge–charge interactions are introduced in the phenyl-hexyl column, leading to a larger retention if compared to MP-1 (19). These changes in both peptide and stationary phase characteristics make interactions more or less favored, leading to a different retention depending on the mobile and stationary phase type.
Effect of Ion-Pair Reagents on Peak Shape and Efficiency
For the sake of comparison, the effect of the ion-pair reagent on the chromatographic behavior of glucagon on the two columns has been investigated and reported in Figure 3.

FIGURE 3: Comparison between chromatograms measured at 31% acetonitrile on (a) C18, and (b) phenyl-hexyl columns with MP-1 (blue) and MP-2 (red). The number of theoretical plates per meter (N/m) and the tailing factor (tF) measured at 5% peak height are indicated for each peak.
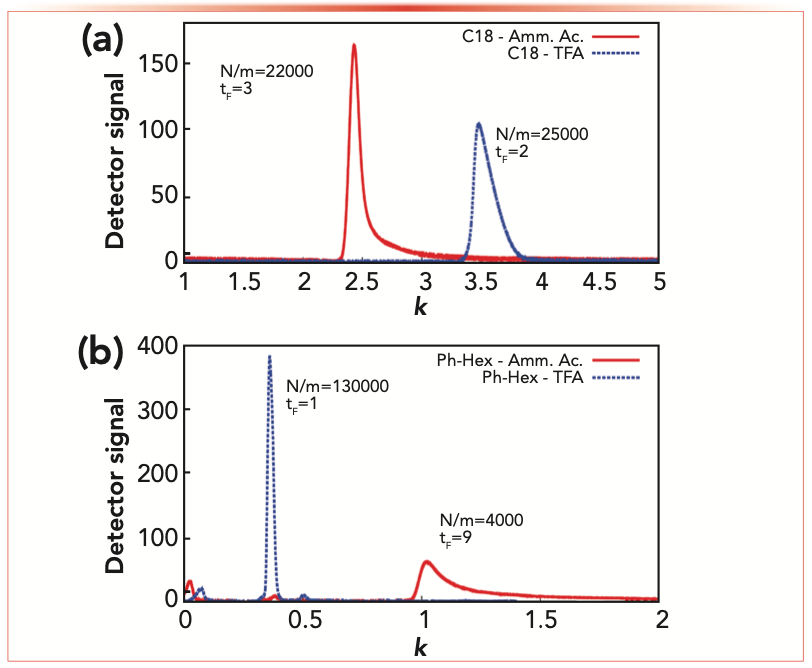
From these plots it can be evinced that the peak shape is highly influenced by the type of ion-pair reagent used. For both columns, ammonium acetate leads to a significant peak tailing (red) compared to TFA (blue). Moreover, the tailing factor measured with MP-2 for the C18 column (tF = 3) is 3 fold smaller with respect to the one measured on the phenyl-hexyl column (tF = 9), indicating that, as already pointed out in the previous section, analyte interactions with residual silanols are much more pronounced on the latter stationary phase. As a result, efficiency measured with MP-2 on the phenyl-hexyl column (N/m = 4000) is three times less than C18 column (N/m = 22000). On the other hand, TFA leads to more Gaussian peaks, especially with phenyl-hexyl stationary phase, and, as a consequence, to efficiencies as high as 130,000 N/m.
These information may be helpful for the selection of the correct combination of stationary and mobile phases for the development of highly efficient and fast analytical methods for the identification and quantification of the target peptide. In this regard, a phenyl-hexyl column in combination with TFA as ion-pair reagent may be an ideal candidate for ultrafast separations.
Conclusions
In this work, retention behavior of a therapeutic peptide, glucagon, has been investigated on two RPLC columns, C18 and phenyl-hexyl, using a binary mobile phase made of acetonitrile and water with two ion-pairing reagents, TFA, and ammonium acetate.
Excess adsorption isotherms have shown that the total amount of mobile phase adsorbed is 20% higher on the phenyl-hexyl column with respect to the C18 one. Acetonitrile adsorption on the phenyl-hexyl column may interfere with π-π interactions between analyte and phenyl groups in the stationary phase, with important consequences on retention. Moreover, it has been demonstrated that the characteristics of both the ion-pair reagent and the ligand type have a deep influence on the type of interaction established between analyte and stationary phase, the peak shape and the efficiency of the separation.
This information is of fundamental importance for the development of reliable, selective, and fast analytical methods able to separate and identify the tar- get peptide.
Acknowledgments
The authors thank the Italian University and Scientific Research Ministry (grant PRIN2017Y2PAB8_003, title: “Cutting edge analytical chemistry methodologies and bio-tools to boost precision medicine in hormone-related diseases”).
References
(1) C. De Luca, S. Felletti, M. Macis, W. Cabri, G. Lievore, T. Chenet, L. Pasti, M, Morbidelli, A. Cavazzini, M. Catani, and A. Ricci, J. Chromatogr. A 1616, 460789 (2020).
(2) C. De Luca, S. Felletti, G. Lievore, A. Buratti, T. Chenet, L. Pasti, M. Morbidelli, A. Cavazzini, M. Catani, M. Macis, A. Ricci, and W. Cabri, J. Chromatogr. Sep. Tech. 11(428), (2020). DOI:10.35248/2157-7064.20.11.428
(3) S. Wegmüller and S. Schmid, Curr. Org. Chem. 18(8), 1005–1019 (2014).
(4) S. Chandrudu, P. Simerska, and I. Toth, Molecules 18(4), 4373–4388 (2013).
(5) S. Bernardi, D. Gétaz, N. Forrer, and M. Morbidelli, J. Chromatogr. A 1283, 46–52 (2013).
(6) V. Sanz-Nebot, F. Benavente, I. Toro, and J. Barbosa, Anal. Bioanal. Chem. 377, 306–315 (2003).
(7) C. De Luca, S. Felletti, G. Lievore, T. Chenet, M. Morbidelli, M. Sponchioni, A. Cavazzini, and M. Catani, Trends in Anal. Chem. 132, 116051 (2020).
(8) B. Bobály, V. Mikola, E. Sipkó, Z. Márta, and J. Fekete, J. Chromat. Sci. 53, 1078–1083 (2015).
(9) T. Müller-Späth, G. Ströhlein, O. Lyngberg, and D. Maclean, Chem. Today 31, 56–61 (2013).
(10) L. Aumann and M. Morbidelli, Biotech. Bioeng. 98, 1043–1055 (2007).
(11) B. Buszewski, Sz. Bocian, and A. Felinger, J. Chromatogr. A 1191, 72–77 (2008).
(12) G. Guiochon, A. Felinger, D.G. Shirazi, and A.M. Katti, Fundamentals and Preparative and Nonlinear Chromatography, 2nd Edition (Academic Press, Elsevier, Amsterdam, The Netherlands, 2006).
(13) N. Marchetti, A. Cavazzini, L. Pasti, and F. Dondi, J. Sci. Sep. 32, 727–741 (2009).
(14) F. Chan, L.S. Yeung, R. LoBrutto, and Y.V. Kazakevich, J. Chromatogr. A 1082, 158–165 (2005).
(15) S. Bocian, P. Vajda, A. Felinger, and B. Buszewski, J. Chromatogr. A 1204, 35–41 (2008).
(16) P. Vajda, A. Felinger, and G. Guiochon, J. Chromatogr. A 1291, 41–47 (2013).
(17) K. Croes, A. Steffens, D.H Marchand, and L.R. Snyder, J. Chromatogr. A 1098, 123–130 (2005).
(18) J.S. Pedersen, J. Diabetes Sci. Technol. 4, 1357–1367 (2010).
(19) M. Gilar, K.J. Fountain, Y. Budman, U.D. Neue, K.R. Yardley, P.D. Rainville, R.J. Russell II, and J.C. Gebler, J. Chromatogr. A 958, 167–182 (2002).
Simona Felletti, Chiara De Luca, Giulio Lievore, Alessandro Buratti, Desiree Bozza, Alberto Cavazzini, and Martina Catani are with the Department of Chemical, Pharmaceutical, and Agricultural Sciences at the University of Ferrara in Ferrara, Italy. Marco Macis and Antonio Ricci are with Fresenius Kabi iPSUM in Villadose, Italy. Walter Cabri is with the Department of Chemistry at the University of Bologna in Bologna, Italy. Direct correspondence to: fllsmn1@unife.it












, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




