The Multi-Attribute Method (MAM) for the Characterization of Biopharmaceuticals


New analytical workflows are needed to address the advances in biopharmaceutical product composition. A description of the multi-attribute method (MAM) is given, which has been developed to monitor critical quality attributes (CQAs) simultaneously and directly.
Over the last decade or so, biopharmaceuticals have gained market share and become more complex in their composition. Therefore, new analytical workflows are needed to address these advances, and one of those workflows that has gained substantial traction over the last several years is the multi-attribute method (MAM). MAM was originally developed to be used throughout the product life cycle, from process development through quality control, and has become even more popular as quality by design (QbD) has become a more prevalent approach for biopharmaceutical development. MAM is designed to monitor critical quality attributes (CQAs) simultaneously and directly, such as sequence, post-translational modifications, and impurities, making it a more streamlined and productive workflow for biopharmaceutical analysis. In this column, we will discuss the role of liquid chromatography and mass spectrometry in MAM, as well as other new technologies and anticipated advances of MAM that are on the horizon.
Throughout biopharmaceutical development, both drug substance and drug product are characterized using a variety of analytical techniques. The analytical techniques that are used range from low resolution gel electrophoresis to high resolution mass spectrometry. Over the last several decades, mass spectrometry has been a staple of research and development as well as process development, and has made its way into quality control (QC) of biologics. The shift in the quality paradigm to a Quality by Design (QbD)-based approach has opened the door to new analytical tools for the analysis of biopharmaceuticals throughout the life cycle of the product, including QC. In this column, we will look at the multi-attribute method as one of those new analytical tools, largely based on mass spectrometry.
Regulatory Landscape and Quality by Design (QbD)
Biopharmaceuticals such as monoclonal antibodies (mAbs) are drugs that are produced by living cells, which gives them an inherent heterogeneity. In all the years of protein analysis it is rare to come across a homogeneous drug product. At the very least, there is heterogeneity in the post-translational modifications (such as deamidation, oxidation, and glycosylation). This complexity has led to a need to monitor biopharmaceuticals throughout their life cycle from manufacture to storage.
Over the last several years, national regulatory authorities have started to more robustly recommend a QbD approach to the manufacture of drug products (4,5). At its core, a QbD approach considers quality of the final marketed drug product from the very first experiment done. Quality is considered and built into the design and development of the drug, leveraging the diversity of expertise needed to do so from day one. To put it another way, quality is considered at the beginning of the process, and is at the heart of the entire drug’s life cycle. In this approach, product quality attributes (PQAs) of the drug product can be understood at the molecular level, ensuring the safety and efficacy of the drug.
From these PQAs, as part of a QbD approach, a quality target product profile (QTPP) is developed, and specific critical quality attributes (CQAs) and implementation of control strategies are identified to monitor and ensure quality, efficacy, and safety (1,2,5–8). For our purposes, PQAs are any attribute of the drug, whether critical or noncritical. On the other hand, a CQA, as defined by the International Council for Harmonisation- Q8 and Q11 guidelines (www.ich.org) is a “physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range or distribution to ensure the desired product quality.” Therefore, CQAs are a subset of PQAs that are critical for the safety, efficacy, or quality of the drug. CQAs are applied to drug substances, excipients, in-process materials, and the drug product. Thus, this product complexity and the QbD approach are developed to provide flexibility while reducing the product life cycle management burden (1).
What is the Multi-Attribute Method?
The need to monitor a drug throughout its life cycle, and the movement to QbD approaches, has led to a need for more advanced analytical tools that are capable of monitoring product quality. Until recently, CQAs had to be monitored using a set of orthogonal analytical techniques, which included immune assays, SDS gel-based approaches, chromatographic approaches, electrophoretic approaches, biophysical approaches, and mass spectrometry. Most of these techniques only monitor one characteristic, and are typically indirect measurements. Many techniques used to monitor small molecules cannot be used, due to limitations of these testing methods for biopharmaceuticals.
At the end of the day, the combination of multiple techniques to monitor CQAs is both time consuming and expensive, leaving the door open for analytical advances that allow for the monitoring of CQAs throughout the products life cycle (8).
As the regulatory landscape evolved to promote QbD, advances in mass spectrometry (MS) also occurred, making MS into one of the most, if not the most, prevalent analytical technique used to characterize biopharmaceutical products. One main advantage of MS was its ability to perform this analysis on multiple attributes in fewer analysis. Therefore, it was only logical that mass spectrometry would be adapted to simultaneously monitor many CQAs, thus leading to an MS-based multi-attribute method (MAM). MAM provides an opportunity to monitor multiple attributes, potentially reducing cost, providing a deeper knowledge of the product, and potentially accelerating the product’s release. It also provides a direct analysis, as opposed to the indirect analysis of more conventional techniques. A single MAM has the potential to monitor 20 unique critical quality attributes (including glycation, glycoslylation, mutations, and host-cell protein profiling) (8) that can facilitate product review and approval at reduced costs.
The Multi-Attribute Method
MAM is a single analytical tool that provides analysis of multiple CQAs, taking advantage of the high-resolution capabilities of certain MS instruments. CQAs are determined through analysis of the product quality attributes. MAM consists of two main components: 1) targeted attribute quantitation at the amino acid level and 2) new peak detection (NPD). Often, the targeted attribute quantitation includes the monitoring of an identified CQA that is a specific post-translation modification (glycosylation or oxidation) or peptide of interest. On the other hand, NPD is a comparative analysis between chromatograms where unexpected changes in the product can be detected by a peak alignment algorithm, essentially an impurity screen. Interestingly, when NPD is not used, instead of multi-attribute method it is referred to as multi-attribute monitoring (1,3).
The most common MAM starts with an enzymatic digest of the biopharmaceutical product. It is important to ensure that the digestion and other steps of the MAM do not introduce artificial modifications (1,9,10). After digestion, a bottom-up MS approach is used; that is, the digested peptides are analyzed using tandem MS ( LC–MS/MS) that originally will be used to identify the product quality attributes to be monitored, including specific post-translation modifications. In addition, these PQAs can be quantitated using extracted ion chromatograms (EICs), enabling the first major component of MAM—targeted attribute monitoring. In addition to PTMs, the analysis also provides sequence coverage of the molecule (Figure 1) (1,3).


FIGURE 1: An example of a MAM workflow.
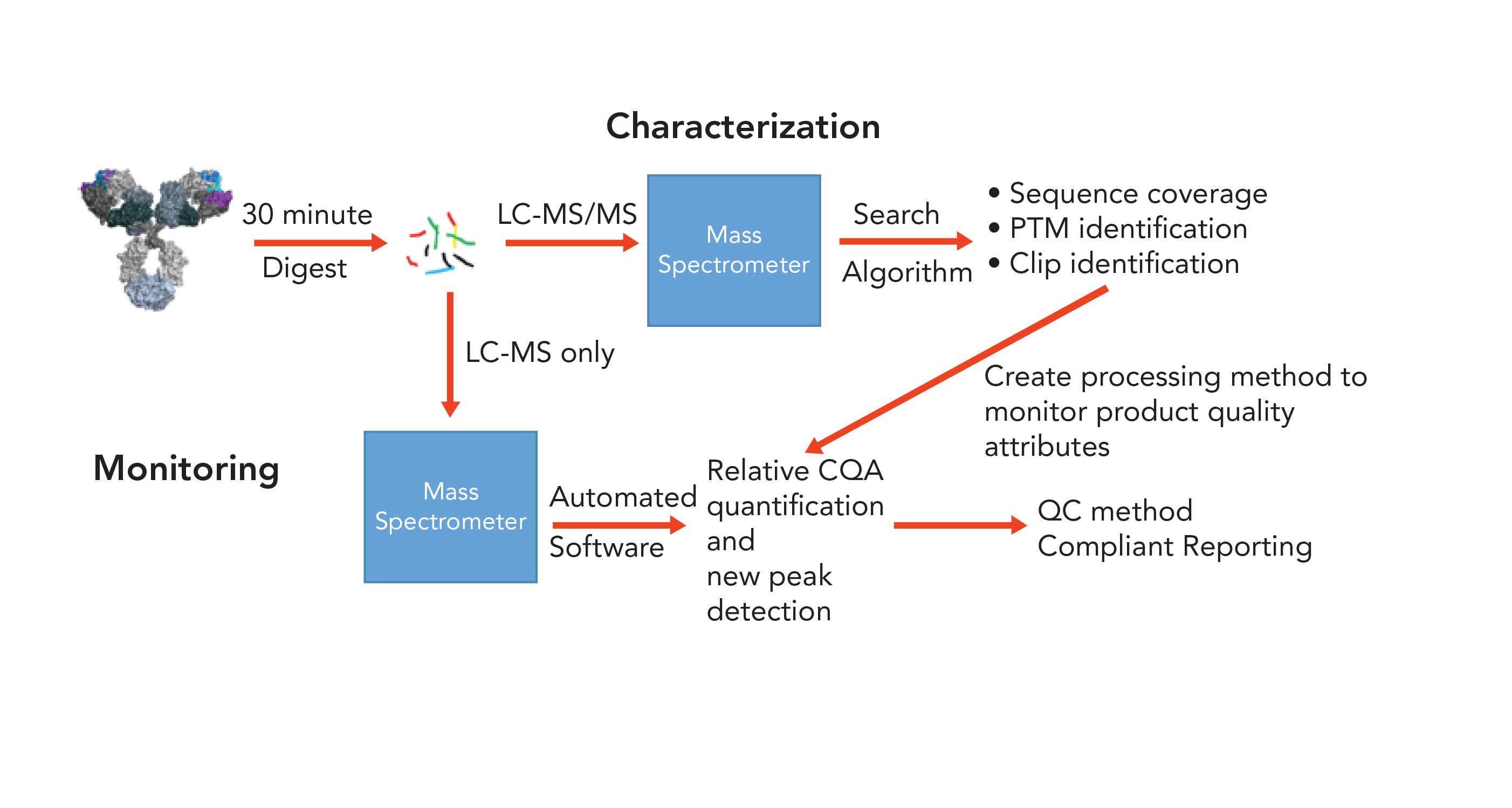
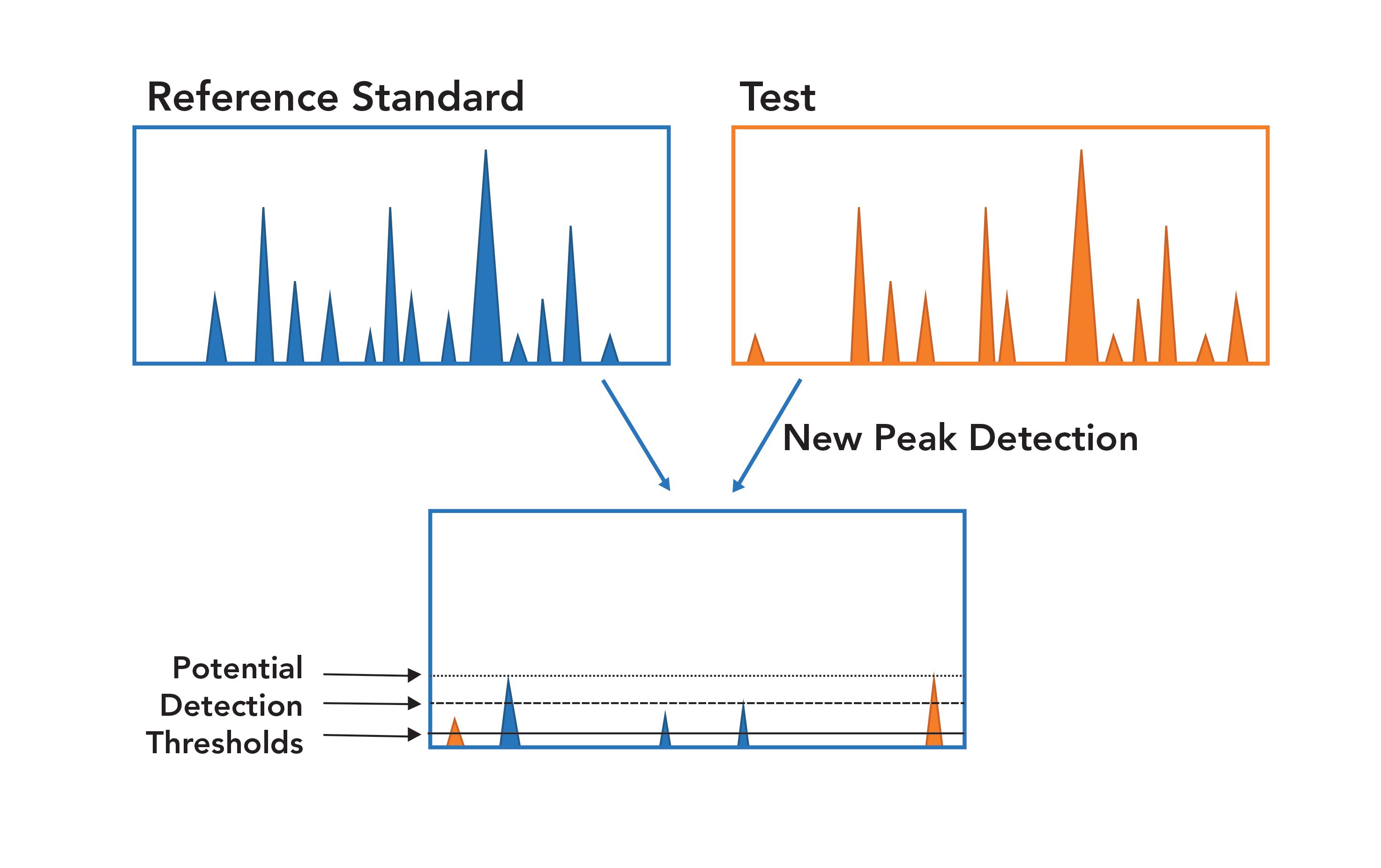
After initial LC–MS/MS analysis, CQAs can be monitored using chromatogram fingerprinting looking for any differences (Figure 1). This enables the new peak detection component of MAM. Specifically, NPD allows for non-specific, new, or lost peak detection through m/z-based alignment of chromatograms. It is important to ensure that the appropriate experimental parameters are set and consistently used, because any changes to these parameters could affect the results. In Figure 2, we see an example of where NPD can be used (Figure 2). The test of an experimental sample (orange) has two peaks not in the reference standard (blue), and is missing three peaks from the reference samples. As highlighted by Figure 2, the detection thresholds for NPD are crucial. If the threshold is too high, differences could be missed, resulting in false negatives; if the threshold is too low, noise maybe be picked up as false positives that may trigger unnecessary further investigations (3).

FIGURE 2: New peak detection (NPD). Reference standard (blue) compared to the test sample (orange); the bottom graph showing new peek’s identified.
Why MAM?
As discussed above, MAM allows for directed monitoring of specific CQAs, which is not only a potential benefit to research and development and process development, but also to the quality control environment. Thus, MAM’s quantitative ability and ability to identify changes in chromatogram peaks makes it an analytical tool well suited to monitor changes in the product during manufacturing, long-term storage, and preparation for clinical use (3).
When considering MAM in the QC environment, one should consider four issues: risk assessment, method validation, capabilities and specification of new peak detection, and comparison to conventional methods. When considering risk assessment, we generally ask what characteristics of the biologic cannot be monitored using MAM, or what characteristic might we miss. For example, degradation details of the biopharmaceutical maybe lost due to the use of a protein digest. Risk assessment is in relation to the relevance of differences or lost information related to quality, safety, and efficacy.
When considering method validation, one must ask themselves if the precision, detection, and quantitation limits of the instruments being used for MAM are sufficient to monitor the CQAs of interest. Not only do we have to consider the instrument capabilities, but also the operator’s capabilities (3). Many MAMs use LC–MS peak monitoring after the initial LC–MS/MS analysis because the operators are not MS experts (2). Do the operators have adequate knowledge of MS to implement a successful MAM? When considering new peak detection, we must ensure appropriate thresholds to avoid detection of noise or missing of relevant peaks. Lastly, when considering MAM, we should ask how MAMs compare to traditional approaches for analysis of biopharmaceuticals (3).
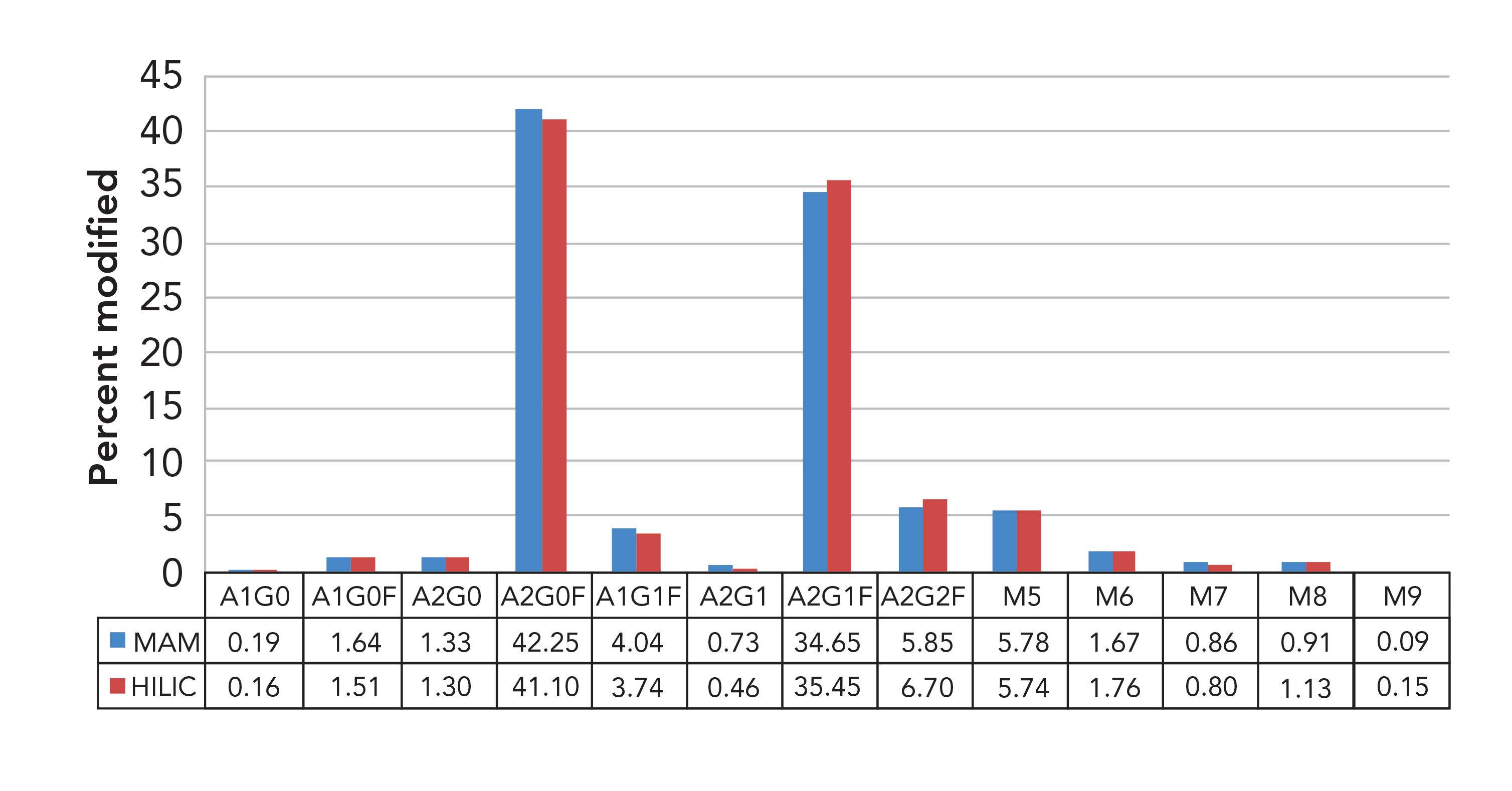
Over the last several years, these analyses have been performed many times, generally showing that MAM is as good as, or better at, detecting and monitoring certain CQAs as other approaches. For example, Figure 3 shows an analysis between the traditional approach, hydrophilic-interaction chromatography (HILIC), to identify one of the most crucial CQAs for a biologic, glycans, versus MAM (Figure 3). In this case, you can see that MAM performed similarly to HILIC (2).

FIGURE 3: Traditional HILIC assay of glycans compared to a MAM glycan analysis. Both assays are comparable.
MAM and Control Strategy
MAM can help improve control strategies and advanced manufacturing. ICH-Q10 (www.ich.org) defines a control strategy as “a planned set of controls, derived from current product and process understanding, that assures process performance and product quality.” Monitoring the product throughout its life cycle allows us to mitigate risk, and to increase the confidence in the control strategy in place throughout the product life cycle. MAM would allow for any deviations or changes to be observed, which is not possible with conventional methods. In addition, MAM quantitatively monitors specific attributes at the molecular level, allowing a direct link between PQAs and clinical performance. This can help establish clinically relevant specifications using direct attribute measurements improving the control strategy (1).
The Future of MAM
MAM is in its early stages of implementation, which has been accelerated by the regulatory paradigm shift in the approach to QbD with the approval and implementation of ICH-Q8 through ICH-Q11. As discussed, MAM allows for monitoring of a biopharmaceutical throughout its life cycle, and with advances in automation (automated sampling) and data analytics, sampling from the bioreactor, followed by sample cleanup, MAM will be able to provide near real-time measurements of the product. In time, MAM will be more routinely considered a tool in our process analytical technology (PAT) toolbox (1,4,5).
As more manufactures provide 21 CFR Part 11 compliant software and GxP compliant instruments, MAM will become more ubiquitous in quality control (QC) laboratories over time (including as a PAT). Also, as more regulators and regulatory authorities become familiar with and comfortable with MAM data, it will be more robustly used to monitor CQAs. Therefore, in the coming years, MAM will be used to assess all molecular attributes in one single analysis in a fully automated fashion, in line with ICH guidelines and local regulatory guidance (2).
Improvements in analytical instrumentation will also enable further uptake of MAM throughout the life cycle of the biopharmaceutical. Currently, LC–MS/MS is the gold standard in peptide mapping, post-translational modification, and peak monitoring, where most MAMs take advantage of bottom-up proteomics. As mass spectrometry methods become more robust at identifying tertiary and quaternary structure, one could envision MAM including some sort of structural analysis, perhaps using ion-mobility mass spectrometry. Another such advancement in MS could be in top-down MS, where protein digest is not needed to analyze the product (2).
Although still in its infancy, MAM has the potential to revolutionize the way we do analytics for biopharmaceuticals. It likely will allow continued monitoring of biopharmaceuticals throughout the life cycle of the product. This is important for traditional monoclonal antibody drugs; however, as the drug products become more complicated, such as in the case of anti- body drug conjugates (ADC), it becomes even more important to have continuous monitoring of the drug product to ensure quality, safety, and efficacy of the drug. Beyond traditional biotherapeutics, personalized or precision medicine could also benefit from MAM (with appropriate modifications to the methodology), as these drug products are developed for one specific patient. As instrumentation becomes more robust, automated, and easier to use, the use of MAM will increase as the operators of instrumentation for these experiments are often not experts. One such advancement in automation may come from advances in artificial intelligence as applied to MAM. Thus, MAM is an analytical technology that will help improve drug quality, safety, and efficacy for the benefit of the patient.
References
(1) R.S. Rogers, M. Abernathy, D.D. Richardson, J.C. Rouse, J.B. Sperry, P. Swann, J. Wypych, C. Yu, L. Zang, and R. Deshpande, AAPS J. 20(7), 1–8 (2018).
(2) R.S. Rogers, N.S. Nightlinger, B. Livingston, P. Campbell, R. Bailey, and A. Balland, MAbs 7(5), 881–90 (2015).
(3) S. Rogstad, H. Yan, X. Wang, D. Powers, K. Brorson, B. Damdinsuren, and S. Lee, Anal Chem. 91(22), 14170–14177 (2019).
(4) A. S. Rathore and H. Winkle, Nat Biotechnol. 27(1), 26–34 (2009).
(5) X. Xu, H. Qiu, and N. Li, Journal of Applied Bioanalysis 3(2), 21–25 (2017).
(6) M. Pathak, D. Dutta, and A. Rathore, Electrophoresis 35(15), 2163–2171 (2014).
(7) A.S. Rathore, Trends Biotechnol. 27(9), 546–553 (2009).
(8) Y. Zhang, J. Guo, Bioanalysis 9(6), 499–502 (2017).
(9) J.R. Auclair, J.P. Salisbury, J.L. Johnson, G.A. Petsko, D. Ringe, D.A. Bosco, N.Y. Agar, S. Santagata, H.D. Durham, and J.N. Agar, Proteomics 14(10), 1152–1157 (2014).
(10) S. Liu, K.R. Moulton, J.R. Auclair, and Z.S. Zhou, Amino Acids 48(4), 1059–1067 (2016).
ABOUT THE CO-AUTHOR


Jared Auclair is an Associate Dean of Professional Programs and Graduate Affairs at the College of Science at Northeastern University, in Boston, Massachusetts. He is also the Director of Biotechnology and Informatics, as well as the Director of the Biopharmaceutical Analysis Training Laboratory.
ABOUT THE COLUMN EDITOR


Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.






 for the Characterization of Biopharmaceuticals")








Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

