The Determination of Trace Per- and Polyfluoroalkyl Substances and Their Precursors Migrated into Food Simulants from Food Contact Materials by Liquid Chromatography–MS/MS and Gas Chromatography–MS/MS
An increasing range of per- and polyfluoroalkyl substances (PFAS) has been found migrating from food contact material into food. This article establishes an integrated analytical approach combining HPLC–MS/MS and GC–MS/MS to detect 36 such PFAS.


Image Credit: Soho A studio/stock.adobe.com

The determination of multiple per- and polyfluoroalkyl substances (PFAS) migrating from food contact material gained in importance as an increasing range of PFAS has been found migrating from food contact material into food. In this study, an integrated analytical approach that combines high performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) and gas chromatography–tandem mass spectrometry (GC–MS/MS) was established for detecting 36 PFAS migrating from food contact materials into four food simulants (3% acetic acid, 10% alcohol, 50% alcohol, and olive oil). The response surface methodology was applied to optimize the selection of solvents in sample treatment. This target analytical method was appropriate for the determination of multiple PFAS, with recoveries ranging from 81.8 to 118.7%. The relative standard deviations (RSDs) ranged from 2.4 to 7.8%, and detection limits were in the range of 0.3 to 10 µg/kg in relevant food simulants.


Per- and polyfluoroalkyl substances (PFAS) are a family of synthetic substances that do not occur naturally in the environment. They are a concern due to their stable physical and chemical properties with strong C-F bonds, including their chemical inertness, thermal stability, high surface activity, and hydrophobic-oleophobic properties (1–3). PFAS are widely used in consumer goods, household products and food contact materials (4–5). In food contact material (FCM), PFAS are mainly used in nonstick cookware, as well as the coatings of paper and other resistant materials, due to their oil- and water-repellent properties (6–7). Studies have indicated that the migration of PFAS from FCM into food is likely to be the main route of consumer exposure to these substances (8–11).
PFAS have been found to be highly resistant, and could persist in the environment for long periods of time, as well as in human serum, milk, and tissues (12–17). Certain type of PFAS, such as perfluorocarboxylic acids (PFCA) and perfluorosulfonic acids (PFSA), are likely to be toxic and bioaccumulate, posing adverse effects on human health (18–21). Perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are two of the most studied PFCA and PFSA. There is suggestive evidence from human epidemiology studies that PFOS and PFOA may cause abnormal liver enzymes, antibody response, and cancer (21–23). To reduce the occurrence of PFOS and PFOA, a number of PFAS, such as perfluoro‑alcohol and its derivatives, have been used as substitutes to replace PFCA and PFSA in FCM. However, studies indicated that these substitutes could also pose health risks to humans, as PFSA and PFCA precursors are more toxic than the PFSA or PFCA themselves (24). In addition, these precursors could be absorbed into the human body, and degrade into PFOA and PFOS by an oxidation mechanism (25). For example, 8:2FTOH (flurotelomer alcohol) and 10:2FTOH could be converted into PFOA, and PFDA, and N-methyl perfluorooctane sulfonamido ethanol (N-MeFOSE) and N-ethyl perfluorooctane sulfonamido ethanol (N-EtFOSE) could be capable of conversion into PFOS. Therefore, besides known harmful PFAS, the migration of PFAS precursors from food contact material must also be taken into account to ensure its safety.

To protect consumers from exposure to PFAS migrated from FCM, stringent regulatory approaches have been adopted to control their production, application, and migration. The Danish Food and Veterinary Administration set a recommended limit for the total organic fluorine content in paper and cardboard at 0.35 μg per square decimeter of paper. The U.S. Food and Drug Administration (FDA) finalized an amendment to regulations that certain type of PFAS were not permitted as additives used in the manufacture of FCM (26). In China, the National Food Safety Standard GB 9685, which is the regulation for the use of additives in FCM, added an amendment in 2016 that no longer authorized PFAS as additives in the manufacture of FCM (27). In 2017, The European Chemicals Agency (ECHA) added seven types of PFCAs and PFSAs to the Substances of Very High Concern (SVHC) list (28), which attracted much attention concerning the occurrence of PFAS, and their effects on the environment and human health.
Comparable results for measurement of PFAS migration from FCM is crucial for official control purposes. To achieve this objective, the guidance for choice of food simulants and migration test conditions for plastics have been provided in relevant regulations (29), which define the food simulants that should be used to mimic a specific foodstuff, and what standardized conditions of time, temperature, and contact should be performed. Various analytical approaches have been investigated for measuring the migration of PFAS from FCM (30–35). GC–MS is usually used for detecting volatile fluorine-containing compounds, such as perfluoroalcohols and perfluoroalcohol acrylates. LC–MS/MS, a highly sensitive and selective tool, was applied to detect more than 10 PFAS that mainly refer to straight‑chain perfluorinated carboxylic acids or perfluorinated sulfonic acids. However, an analytical approach that is suitable for determination of multiple PFAS to detect migration from food contact materials has not been well established, especially for the increasing range of precursors, including PFCAs and PFSAs. In addition, the previous studies mainly focused on measuring the residue of PFAS in different matrices of FCM, such as extracts of paper. Few studies have been carried out on detecting migration of PFAS by using conventional simulants that represent the specific foodstuff.

To meet official control purposes, this present study aims to establish an effective method for simultaneously measuring multiple PFAS migrated from FCM, and possibly containing both PFAS and their precursors, including fluorinated carboxylic acid, hydrogen‑substituted fluorinated carboxylic acid, and hydrogen‑substituted fluorinated alkyl acid amides. To achieve this objective, the optimization of sample treatment was carried out by using a response surface methodology. An integrated analytical approach of combining high performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) and gas chromatography–tandem mass spectrometry (GC–MS/MS) was established in four conventional food simulants (3% acetic acid, 10% alcohol, 50% alcohol, and olive oil) that are considered to represent specific foodstuffs.
Experimental
Reagents and Materials
Ultrapure water was purified using a Milli-Q system (Millipore, Milford, Massachusetts, USA). Alcohol and methanol (HPLC‑grade) were purchased from TEDIA Company. Ammonium acetate and acetic acid were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, China), and olive oil was purchased from Sinopharm (Shanghai, China).
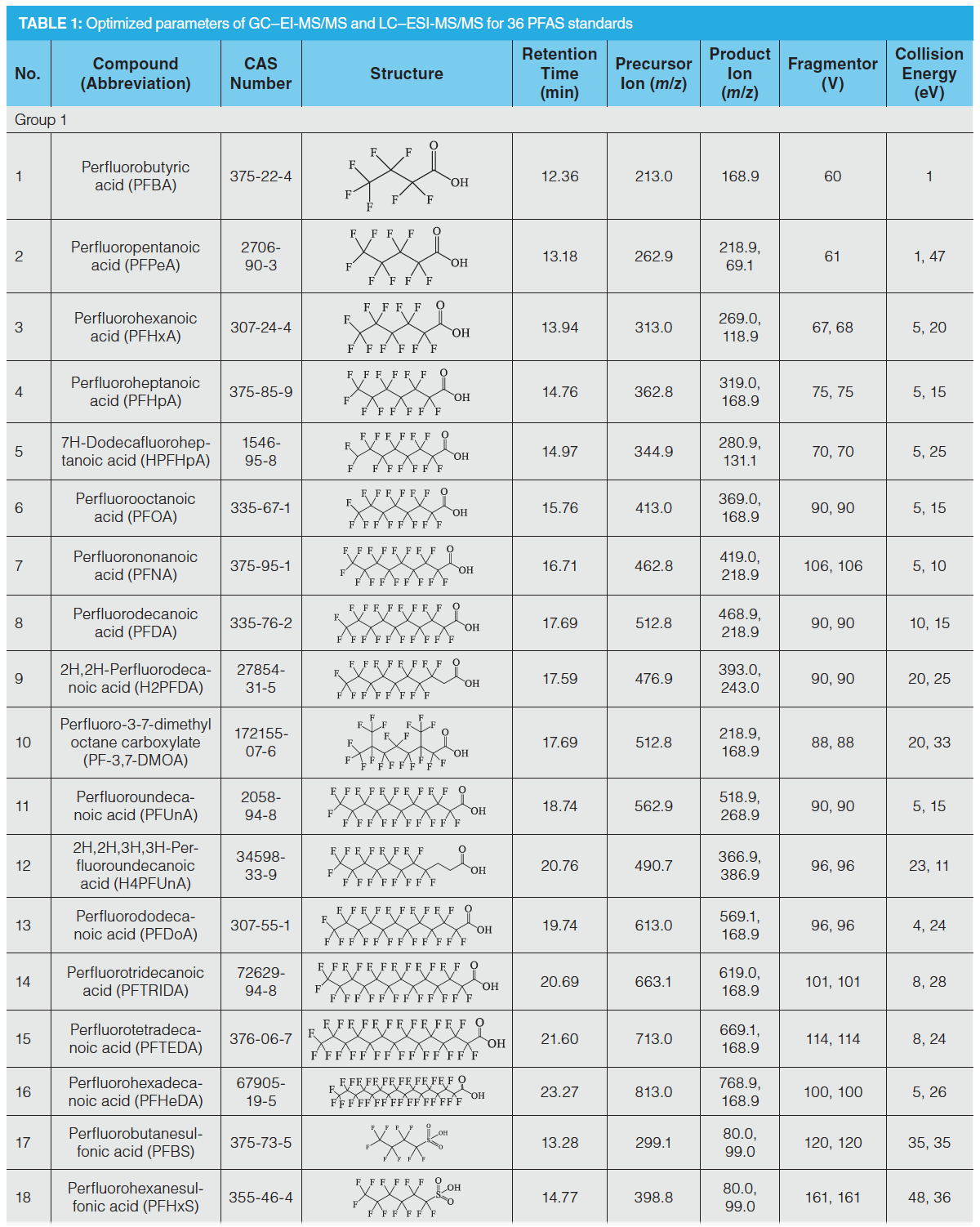
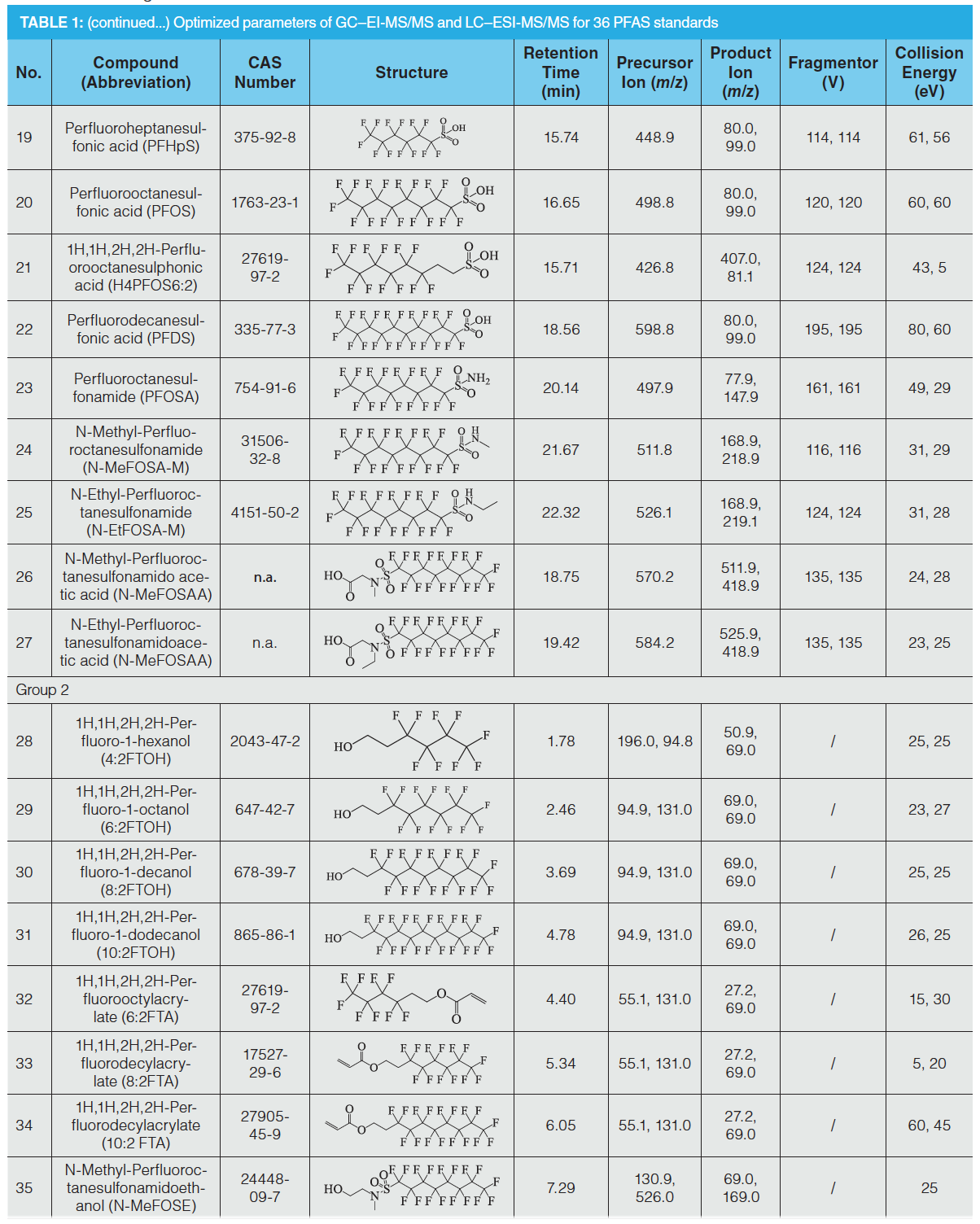
A total of 36 PFAS standards (Table 1) were purchased from Wellington Laboratories (Guelph, Ontario, Canada), Dr. Ehrenstorfer Company (Augsburg, Germany), Chiron Company (Trondheim, Norway), and TRC Company (North York, Ontario, Canada). An intermediate standard solution containing 27 PFAS (0.1 μg/mL, Group 1) was prepared by dissolving the commercial standards in methanol. An intermediate standard solution containing nine PFAS standards (1.0 μg/mL, Group 2) was prepared by dissolving the commercial standards in dichloromethane.



The standard working solutions used for LC–MS/MS analysis were prepared by transferring the intermediate standard (1.0 μg/mL), containing 27 PFAS (Group 1), into a 10 mL volumetric flask, spiked with 50 μL (1.0 μg/mL) 1,2,3,4-13C4 perfluorooctanoic acid (MPFOA) of internal standard, and then made up to 10 mL with the food simulants (10% [v/v] ethanol, 3% [w/v] acetic acid, and 50% [v/v] ethanol), respectively. The standard working solutions used for GC–MS/MS analysis were prepared by transferring the intermediate standard (1.0 μg/mL) containing nine PFAS (Group 2) into a 10 mL volumetric flask, spiked with 50 μL (0.2 μg/mL) methyl margarate-d33 of internal standard, and then made up to 10 mL with the food simulants (10% [v/v] ethanol, 3% [w/v] acetic acid, and 50% [v/v] ethanol), respectively. Then, 2 mL dichloromethane was added, vortexed for 5 min, allowed to separate, and the lower solvent layer was collected for further analysis.
The standard working solutions for the food simulant (olive oil) were prepared by transferring the intermediate standard of Group 1 (0.1 μg/mL) and Group 2 (1.0 μg/mL) into 10 g olive oil, spiked with internal standard of 10 μL MPFOA (1.0 μg/mL) and 50 μL (0.2 μg/mL) methyl margarate-d33. Then, 2 mL acetonitrile was added, vortexed for 5 min to allow stratification, and the upper solvent layer was taken for further analysis.
Equipment
An LC–triple quadrupole mass spectrometer and a GC–triple quadrupole mass spectrometer (6410 Triple Quad LC–MS, 7000c Triple Quad GC–MS, Agilent Technologies, Palo Alto, California, USA) equipped with automatic injectors, were employed for the identification and quantification of PFAS.
The LC column employed was a 15 cm Poroshell120 EC-C18 column (2.7‑µm particle diameter and 2.1‑mm i.d., Agilent Technologies), while the GC column employed was 30 m DB-5 column (0.25‑µm particle diameter and 0.25‑mm i.d., Agilent Technologies). The LC mobile phase consisted of water (solvent A) and HPLC grade methanol (solvent B). The gradient elution procedure was as follows: 0–3 min, 10% B; 3–4 min, 20% B; 4–5 min, 45% B; 5–11 min, 70% B; 11–18 min, 85% B; 18–19 min, 100% B; 19–20 min, 75% B; 20–21 min, 50% B; 21–24 min, 20% B; 24 min, 10% B. The injection volume was 5 μL, and the flow rate was 0.2 mL/min. ESI was used in negative mode with the following conditions: spray ion voltage, 4000 V; nebulizer, 20 psi; gas flow, 8 L/min; and capillary temperature, 350 °C. The GC temperature program was as follows: 75 °C for 3 min, 30 °C/min to 250 °C for 0 min, 50 °C/min to 300 °C for 6 min. The injection volume was 2 μL and pulsed splitless. The gas flow was 1.5 mL/min, with an electron ionization (EI) source and multiple reaction monitoring (MRM) mode. All of the ionization parameters and collision cell parameters were optimized for each analyte (Table 1).
Migration Experimental
Migration experiments were carried out in accordance with European Union (EU) regulation 10/2011 for plastic materials and articles intended to be in contact with food (29).
Each sample was exposed to the food simulants with a ratio of contact area‑to‑volume at 1000 mL:6 dm2, and then placed in an incubator at 70 °C. The food simulants were collected from each sample at 2 h, followed by cooling at room temperature, and then further analysis.
Instrumental Analysis
The majority of PFAS being investigated are fatty acid compounds that can ionize protons, and negatively charged parent ions in aqueous solution. For this reason, LC–MS/MS was used to detect 27 of this type of PFAS (Group 1). For the rest of the nine PFAS being investigated (Group 2), these are generally considered volatile compounds in which perfluoroalcohols and perfluoroalcohol acrylates would not easily be ionized in aqueous solutions; these compounds are more susceptible to ionization in an EI source than in an ESI source, therefore GC–MS/MS was preferred for their analysis.
The PFAS analytes were extracted from the relevant food simulant collected following an exposure step. For the determination of 27 PFAS (Group 1), the food simulants (10% ethanol, 3% acetic acid, and 50% ethanol) were transferred into a 10 mL volumetric flask, and filtered by a 0.22 μm syringe filter, followed by direct LC–MS/MS analysis.
The aqueous simulants containing nine PFAS (Group 2) were extracted with dichloromethane, and a volume of 10 mL of aqueous food simulants was transferred into the headspace bottle, with 50 μL (0.2 μg/mL) methyl margarate-d33 added as the internal standard. Dichloromethane was then added, and vortexed for stratification. The bottom layer was filtered, and used for GC–MS/MS analysis.
For treatment of the olive oil, 10 g of olive oil was transferred into the headspace bottle, and 10 μL (1.0 μg/mL) MPFOA and 50 μL (0.2 μg/mL) methyl margarate-d33 were added as the internal standards. After acetonitrile was added and vortexed for stratification, the supernatant liquor was filtered and analyzed by LC–MS/MS and GC–MS/MS.
Results and Discussion
The Choice of Mobile Phase
A mixture of 27 PFAS comprising a standard at a concentration of 100 ng/mL was used to check the mass spectrometric response and separation power of four different mobile phases, including methanol–water solution, methanol–10 mmol/L ammonium acetate solution (pH = 7), methanol–(containing 0.1% ammonia) water solution, and acetonitrile–10 mmol/L ammonium acetate solution.
The results indicated that the weakest MS signal for target analytes was observed for acetonitrile–10 mmol/L ammonium acetate solution, and the strongest MS signal was observed for methanol–aqueous solution (containing 0.1% ammonia). This is likely due to the MS signal of PFAS analytes being related to the formation of the negative parent ion of PFAS, and the degree of ionization of the proton as well. Methanol allows analytes to generate target ions more easily, and be atomized on the electrospray ionization process, while acetonitrile may decrease the ionization of analytes. The poor peak shapes and baseline disturbances were observed for certain analytes, such as PFBA and PFPeA when methanol–water (containing 0.1% ammonia) was applied. Therefore, methanol–water (containing 0.1% ammonia) was not chosen as the mobile phase for further experiments.
Either methanol–water solution or methanol–10 mmol/L ammonium acetate solution was considered satisfactory as a suitable mobile phase, given that a similar ion peak response was obtained, and good chromatographic peak shapes were observed. It can be explained that the ionization of a proton occurs more often in a neutral or weak alkaline mobile phase, leading to an increase of formation of parent ions. Considering the methanol–water solution has the advantage of making maintenance of instrument pipelines simpler and more effective, the methanol–water solution was preferred for further experiments.
The Choice of Chromatography Column
Three different LC columns, including Agilent Zorbax SB-C18 (150 mm × 4.6 mm, 5‑μm), Poroshell 120 EC-C18 (150 mm × 3.0 mm, 2.7‑µm), and Eclipse XDB C-18 (150 mm × 4.6 mm, 3.5‑µm), were compared in the separation of a mixture of standards (27 PFAS), at a concentration solution of 100 ng/mL.
The results indicated that all three columns could effectively separate the target analytes, particularly n-alkane PFAS. However, due to the occurrence of a high number of target PFAS analytes, the chromatographic peaks of various analytes may overlap each other, causing difficulty in further analysis. The Poroshell 120 EC-C18 was shown to be able to completely separate 27 PFAS analytes in 25 min with the same gradient elution program. Therefore, the Poroshell 120 EC-C18 was chosen as the stationary phase for further experiments.
For the selection of a GC column, baseline separations could be achieved for nine target PFAS using a low or medium polarity column. Therefore, the DB-5, DB-17, or DB-35 column could be chosen to perform the analysis.
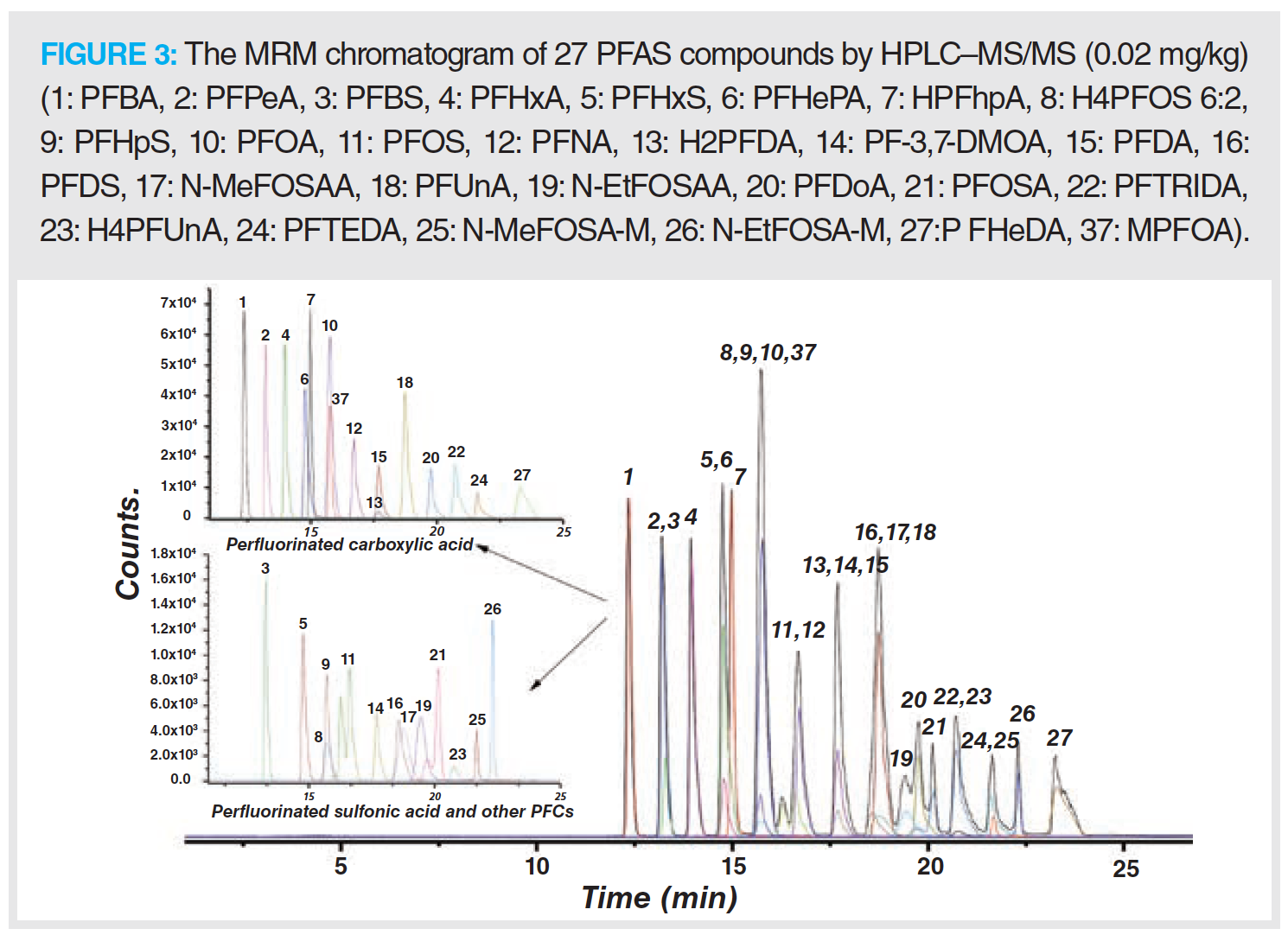
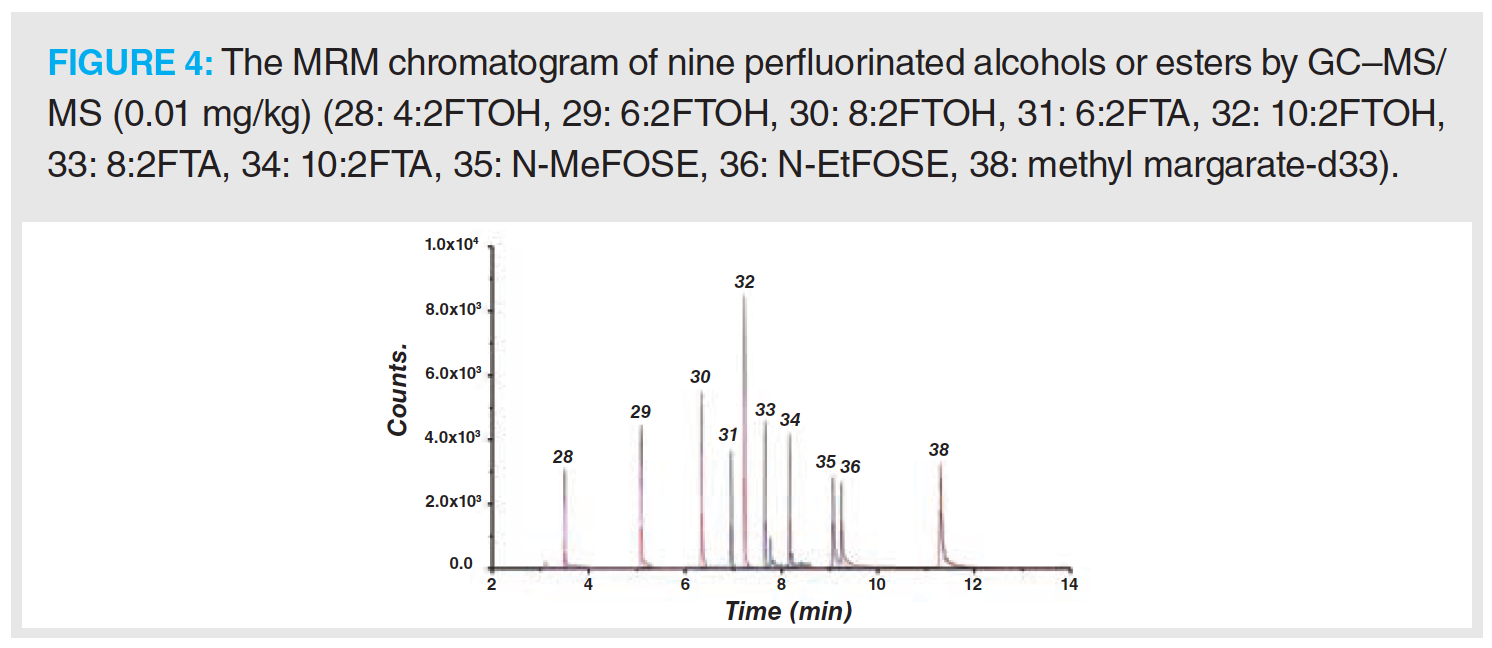
The MRM chromatogram of 36 PFAS compounds is shown in Figures 3 and 4.


The Choice of Extraction Solvent for Olive Oil
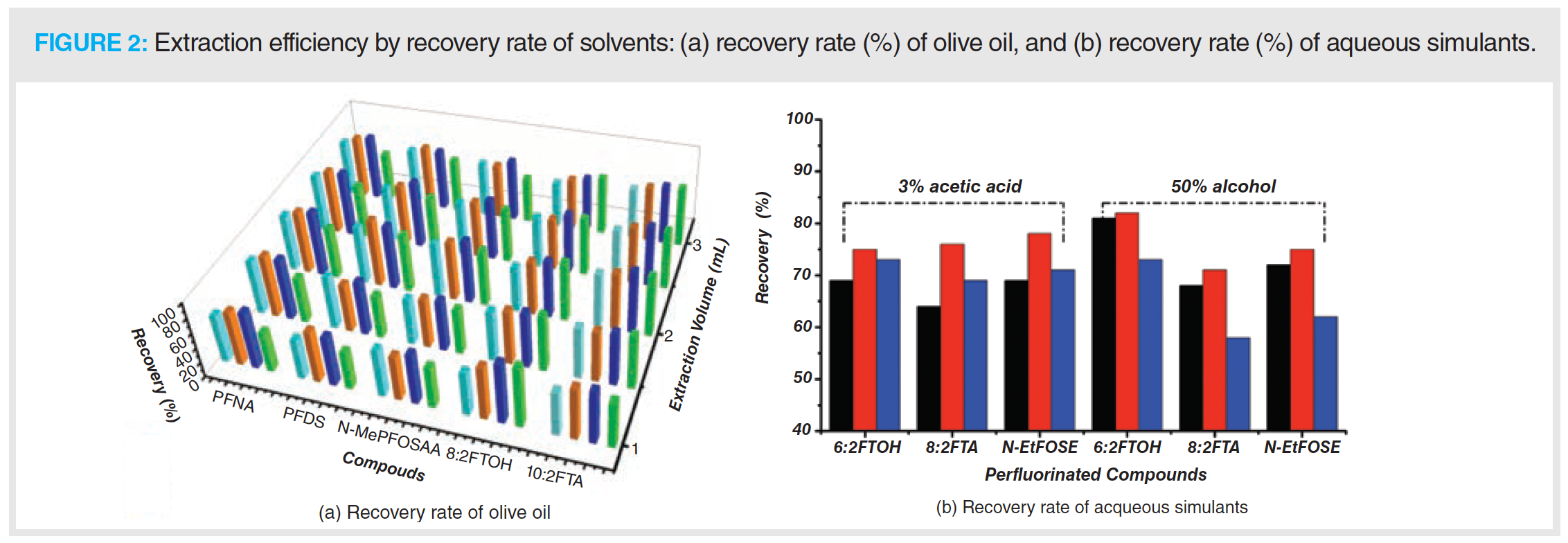
Olive oil, as a fatty simulant, cannot be directly analyzed due to interference from compounds it contains. Therefore, it must be converted into a suitable solution before analysis. To achieve the objective, a recovery experiment was conducted in an olive oil blank spiked with 0.1 mL PFAS standards (10 μg/mL), aimed at selecting a suitable solvent to extract the PFAS from the olive oil. Four different solvents—methanol, acetonitrile:water (1:1), acetonitrile:water (3:1), and acetonitrile—with serial extraction volumes (1 mL, 1.5 mL, 2 mL, 2.5 mL, 3 mL), were investigated. The recovery rate for each solvent is indicated in Figure 2a.
The results indicated that acetonitrile:water (3:1) and acetonitrile had the highest extraction efficiency, and a slightly higher recovery rate was obtained for acetonitrile compared with that of acetonitrile:water (3:1). One of the reasons might be that the PFAS being investigated are long‑chain aliphatic compounds, and the pure organic phase is more favourable for the extraction of the target analytes. For methanol, the separation of the methanol and olive oil layers was not clearly observed after mixing because of the emulsification effect, although the fluorinated fatty alcohol and some perfluorinated carboxylic acids had the highest recovery rates. In the case of acetonitrile:water (1:1) solvent, delamination was not clear, and the recovery rate was low, therefore acetonitrile:water (1:1) was not suitable for GC–MS analysis. Given the test results indicted, acetonitrile was chosen as the solvent to extract the PFAS from the olive oil.
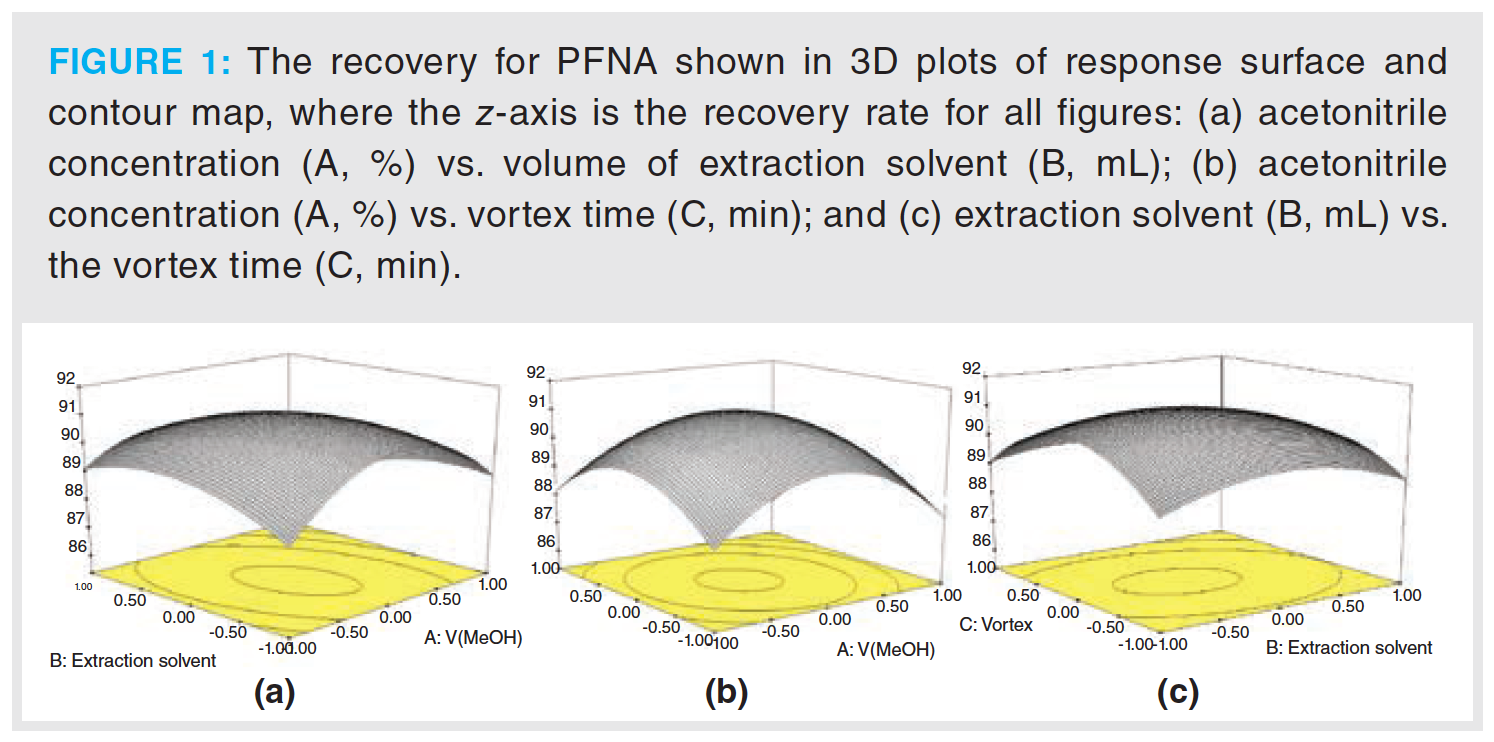
The extraction conditions of PFAS in olive oil were optimized by applying a single‑factor experiment derived from the response surface methodology. Perfluorononanoic acid (PFNA) was taken as the sample to be investigated, and the acetonitrile concentration (A, %), volume of extraction solvent (B, mL), and the vortex time (C, min) were assigned as independent variables. The recovery rate of PFAS were taken as the response value based on the principle of the Box-Behnken central composite design. A secondary polynomial regression model was obtained from 15 experiments with three factors and three levels: recovery = 72.46 + 7.77 × A + 1.47 × B + 5.23 × C − 1.35 × A × B + 0.50 × A × C + 1.75 × B × C + 3.70 × A2 + 0.60 × B2 + 0.69 × C2. The results of the recovery of PFNA obtained by the response surface methodology are shown in Table 2 and Figure 1.


It can be found from the F-value (Table 2) that the sequence of influence of a single factor on the recovery rate was A > C > B, suggesting that the influence of A2 was significant in the quadratic term, while B2 and C2 were not. Thus, 100% acetonitrile was considered an appropriate extraction solution. The highest extraction efficiency was observed when the extraction time was 5 min, and the extraction volume was 2 mL.
The Choice of Extraction Solvent for Aqueous Simulants
In this study, GC–MS was employed for detecting fluorinated fatty alcohols, fluorinated fatty alcohol acrylates, and fluoro-sulfonamide fatty alcohols in the extraction of aqueous simulants. A comparison experiment was conducted on two aqueous simulants (3% acetic acid and 50% ethanol) to check the extraction efficiency of the solvents n-hexane, dichloromethane, and ethyl ether:n-heptane (1:1).
The results showed that all selected solvents were well able to extract the analytes of the aqueous simulants. However, n-hexane was not a suitable solvent to extract fluorinated fatty alcohols, due to its weak polarity. A mix of solvents consisting of ethyl ether and n-heptane did not demonstrate sufficient extraction efficiency. In addition, ethyl ether may cause a hazard to the analyst during vortexing at high pressures. The data indicated that the highest recovery rate was obtained when dichloromethane was applied for measuring fluorinated fatty alcohols and fluorinated fatty alcohol acrylates, which may be due to its medium polarity and strong density that is lower than water‑based solutions. Thus, dichloromethane was chosen as the extraction solvent. Figure 2b shows the extraction efficiency for three different solvents by measured recovery rate. The highest extraction efficiency was achieved with a 5 min extraction time with a 2 mL extraction volume.
Optimization of Mass Spectrometry
The mass spectrometry methods for 27 PFAS in Group 1 and 9 PFAS in Group 2 were optimized to directly detect each PFC (1 mg/L) in methanol. Standard solutions were scanned by performing a first‑stage mass spectrometry full scan in negative ion ESI mode with corresponding transmission voltages. The results showed that the strongest peaks of the perfluorocarboxylic acid, hydrogen substituted fluorocarboxylic acid, or perfluorosulfonamide compounds were the quasi‑molecular ion peaks [M-1]– or the molecular ion peaks following the loss of the carboxyl group [M-45]–. The strongest signal of the perfluorosulfonic acid or hydrogen substituted perfluorosulfonic acid compounds observed were quasi‑molecular ion peaks [M-1]– and sulfonic acid group molecular ion peaks [SO3]–. Nine types of PFAS from Group 2 were scanned by conducting a first‑stage mass spectrometry full scan under the positive EImode. The results indicated that the fluorinated fatty alcohols and fluorinated fatty alcohol acrylates formed smaller fragmentary ion peaks [C3H5F2O]+ (M = 95), (C3H3O]+ (M = 55) or [C3H3F4O]+ (M = 131), which are shown in Figures 3 and 4.
Ion beam scanning was performed with a collision energy of 0–60 eV targeting each quasi‑molecular ion to obtain the daughter ion information and the collision energy values that were suitable for the daughter ion response sizes. Daughter ions of perfluorocarboxylic acid compounds are a series of molecular ions following the loss of the carboxyl group, and there are several CF2 fragments between each molecule, including the ionic fragment of [C3F7]–, [C4F9]–, [C5F11]–, having a general structural formula of [CnF2n+1]–. The ions of the perfluorosulfonic acid compounds were those molecular ions that lost several [CnF2n+1]– saturated carbon chain groups and had sulfonic acid functional groups, such as [CnF2nSO3]–, [SO3]–, and [FSO3]– ions. Both the fluorinated fatty alcohols and fluorinated fatty alcohol acrylates contained the ion-pair [C3H3F4O]+ (M = 131) to [CF3]+ (M = 69). Therefore, the fragment ions with the largest response and least interference were chosen as the quantitative and qualitative ions. To obtain better sensitivities, the parameters including the selection of daughter ions, fragmentation voltage, and collision energy were optimized (see Table 1).
Quantification and Validation
Linear Range and Quantitative Limit
Using the method described above, the 36 PFAS compounds were analyzed in the MRM mode. The concentration and peak area of each compound was taken as the abscissa and ordinate, and a standard curve was drawn. The linear range of the recovered curve was between 1 and 1000 ng/mL, and the limits of quantification (10 S/N) were 0.35–9.72 μg/kg in 3% acetic acid, 0.29–8.43 μg/kg in 10% alcohol, 0.28–8.73 μg/kg in 50% alcohol, and 0.43–9.86 μg/kg in olive oil.
Recovery and Precision
An experiment was carried out where samples were spiked with 10 and 100 ng standards in 3% acetic acid, 10% alcohol, 50% alcohol, and olive oil, respectively. The recovery rates for the four different food simulants ranged from 81.8 to 118.7%, and the relative standard deviations (RSDs) ranged from 2.4 to 7.8%.
Analysis of Real Samples
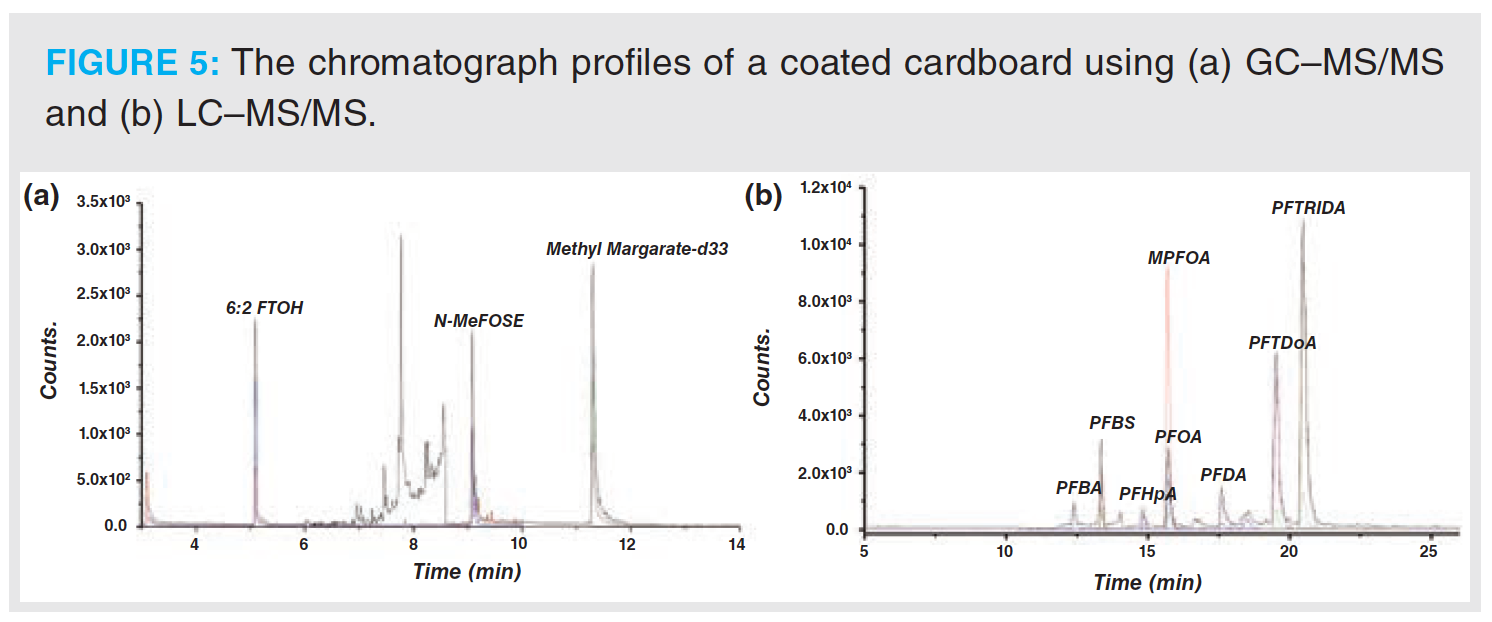
A total of 70 food contact materials were collected from local supermarkets, which include coated paper board, multilayer paper packaging, multilayer plastic food packaging, heat‑resistant rubber articles, and coated heat‑resistant metallic containers. The novel analytical approach was applied to perform the analysis. Some specific PFAS compounds were found in paper samples with concentrations of 0.01 mg/kg to 0.05 mg/kg of food simulants, which accounted for 5% of the total number of samples analyzed. Multilayer cardboard and coated paper board were the main types of packaging in which PFAS compounds were found with average concentrations of 0.02 mg/kg. The highest migration of PFAS was 0.08 mg/kg. The type of PFAS compounds found were mainly straight‑chain perfluorinated acids and perfluorinated alcohol compounds. The chromatogram profiles of the PFAS in typical samples are shown in Figure 5.

Conclusion
A target approach was established for the determination of 36 PFAS migrating from food contact material into food simulants of 3% acetic acid, 10% alcohol, 50% alcohol, and olive oil. A total of 36 PFAS were divided into two groups, and measured by LC–MS/MS and GC–MS/MS. LC–MS/MS was used to detect 27 fatty acid PFAS, while 9 PFAS with volatile properties, including fluorinated fatty alcohols and fluorinated fatty alcohol acrylates were measured by GC–MS/MS. The response surface methodology was a useful tool to simplify the selection of solvents with optimized conditions. This integrated analytical approach was appropriate for the determination of multiple PFAS with recovery rates that ranged from 81.8 to 118.7%, the RSD ranged from 2.4 to 7.8%, and the detection limits ranged from 0.35 to 9.72 μg/kg in 3% acetic acid, 0.29 to 8.43 μg/kg in 10% alcohol, 0.28 to 8.73 μg/kg in 50% alcohol, and 0.43 to 9.86 μg/kg in olive oil. This target approach had the advantage of simultaneously measuring the migration of multiple PFAS from food contact materials with satisfactory sensitivity, accuracy, and reliability. The test results showed that PFAS were found in coated paper and board at mg/kg levels, suggesting that further investigation is needed for the migration of PFAS from coated paper and board.
Funding
The authors acknowledge funds given by national quality infrastructure program (No. 2016YFF0203705), and the General Administration of Quality Supervision Inspection and Quarantine Science-Technology Programs (Grant 2014IK078, Grant 2015IK049). The authors have declared no conflict of interest.
References
- F. Kissa, Fluorinated Surfactants and Repellents (Marcel Dekker Inc., New York, 2nd ed., 2001).
- M.P. Krafft and J.G. Riess, Chemosphere129, 4–19 (2015).
- M.M. Schultz, D.F. Barovsky, and J.A. Environ. Eng. Sci. 20, 487–501 (2003).
- R. Renner, Environ. Sci. Technol. 35, 154A–160A (2001).
- J. Jeon, K. Kannan, H.K. Lim, H.B. Moon, and S.D. Kin, Environ Toxicol.29, 2529–2535 (2010).
- T.H. Begley, K. White, P. Honigfort, M.L. Twaroski, R. Neches, and R.A. Walker, Food Addit. Contam. 22(10), 1023–1031 (2005).
- DuPont. Information on PFOA.2008. http://repanet.de/PFOA2/en_US/index.html. April 07. 2008.
- L.S. Haug, C. Thomsen, and G. Bechert, Environ. Sc. Technol. 43, 2131–2136 (2009).
- X. Trier, K. Granby, and J.H. Christensen, Environ. Sci. Pollut. Res. 118, 108–1120 (2011).
- M.-P. Martinez-Moral and M.T. Tena, Talanta101, 104–109 (2012).
- E. Zafeiraki, D. Costopoulou, I. Vassiliadou, E. Bakeas, and L. Leondiadis, Chemosphere4, 169–176 (2014).
- G.W. Olsen, K.J.Hansen, L.A. Stevenson, J.M. Burris, and J.H. Mandel, Environ. Sci. Technol.37(5), 888–891 (2003).
- K. Inoue, F. Okada, R.S. Kato, S. Sasaki, S. Nakajima, A. Uno, Y. Saijo, F. Sata, Y. Yoshimura, R. Kishi, and H. Nakazawa, Environ. Health Perspect. 112(11), 1204–1207 (2004).
- G.W. Olsen, H-Y Huang, K.J. Helzlsouer, K.J. Hansen, J.L. Butenhoff, and J.H. Mandel, Environ. Health Perspect. 113(5), 539–545 (2005).
- M.K. So, N. Yamashita, S. Taniyasu, Q. Jiang, J.P. Giesy, K. Chen, and P.K.S. Lam, Environ. Sci. Technol. 40(9), 2924–2929 (2006).
- L. Liu, J. She, X. Zhang, J. Zhang, M. Tian, Q. Huang, S.A. Musstjab, A.S. Eqani, and H. Shen, J. Sep. Sci.38, 247–253 (2015).
- T. Zhang, H.W. Sun, Q. Wu, X.Z. Zhang, S.H. Yun, and K. Kannan, Environ. Sci. Technol. 44(9), 3572–3579 (2010).
- C. Lau, J.R. Thibodeaux, R.G. Hanson, M.G. Narotsky, J.M. Rogers, A.B. Lindstrom, and M.J. Strynar, Toxicol. Sci. 90(2), 510–518 (2006).
- N. Johansson, P. Eriksson, and H. Viberg, Toxicol. Sci.108, 412–418 (2009).
- Y. Lin and J. Li, Environ. Chem. 34, 649–655 (2015).
- EFSA (European Food Safety Authority), EFSA Journal653, 1–131 (2008).
- EPA, Health Effects Support Document for Perfluorooctanoic Acid (PFOA), U.S. Environmental Protection Agency EPA822R16003. https://www.epa.gov/sites/production/files/2016-05/documents/pfoa_hesd_final-plain.pdf. (May, 2016; accessed November 17, 2020).
- EPA, Health Effects Support Document for Perfluorooctane Sulfonate (PFOS), U.S. Environmental Protection Agency EPA822R16002. https://www.epa.gov/sites/production/files/2016-05/documents/pfos_hesd_final_508.pdf. (May, 2016; accessed November 17, 2020).
- I. Zabaleta, E. Bizkarguenaga, and D. Bilbao, Talanta152, 353–363 (2016).
- M.J.A. Dinglasan, Y. Ye , E.A. Edwards, and S.A. Mabury, Environ. Sci Technol. 38(10), 2857–2864 (2004).
- FDA, Indirect Food Additives, Paper and Paperboard Components, https://www.federalregister.gov/articles/2016/01/04/2015-33026/indirect-food-additives-paper-and-paperboard-components (2016).
- GB 9685-2016, Standard for the Use of Additives in Food Contact Materials and Articles, http://www.nhfpc.gov.cn/sps/s7891/201611/06ed87a09dad4cf6aee
48cd89efbef35.shtml (2016). - ECHA, Background Document to the Opinion on the Annex XV Dossier Proposing Restrictions on Perfluorooctanoic Acid (PFOA), PFOA Salts and PFOA-related Substances, ECHA/RAC/RES-O-0000006229-7002/F, ECHA/SEAC/RES-O-0000006229-70-03/F (2015).
- Commission Regulation (EU) No. 10/2011 of 14 January 2011 on Plastic Materials and Articles Intended to Come into Contact with Food, https://www.fsai.ie/uploadedFiles/Reg10_2011.pdf (2011).
- W.M. Henderson, E.J. Weber, S.E. Duirk, J.W. Washington, and M.A. Smith, J. Chromatogr. B. 846, 155–161 (2007).
- M. Stadalius, P. Connolly, K.L. Empereur, J.M. Flaherty, T. Isemura, M.A. Kaiser, W. Knaup, and M. Noguchi, J.Chromatogr. A. 1123, 10–14 (2006).
- F. Gosetti, U. Chiuminatto, D. Zampieri, E. Mazzucco, E. Robotti, G. Calabrese, M.C. Gennaro, and E. Marengo, J. Chromatogr. A. 1217, 7864–7872 (2010).
- A.S. Lloyd, V.A. Bailey, S.J. Hird, A. Routledge, and D.B. Clarke, Rapid Commun. Mass Spectrom. 23, 2923–2938 (2009).
- S. Poothong, S.K. Boontanon, and N. Boontanon, Hazard Mater. 205–206, 139–143 (2012).
- B.M. Surma , W. Wiczkowski, H. Zieliński, and E. Cieślik, Packag. Technol. Sci. 28, 789–799 (2015).
Dan Li, Zi-hao Zhang, Huai-ning Zhong, Jing-jing Pan, Jian-guo Zheng and Hui Liu are with the Guangdong Inspection and Quarantine Technology Centre and the Guangdong Provincial Key Laboratory of Import and Export Technical Measures of Animal, Plant and Food, in Guangzhou, China. Lei Zhu is with the China National Center for Food Safety Risk Assessment, in Beijing, China. Qin-bao Lin is with the Department of Food Science and Engineering at Jinan University, in Guangzhou, China. Direct correspondence to: zhulei@cfsa.net.cn or marco_zhong@iqtc.cn or to the Editor-in-Chief, amatheson@mjhlifesciences.com








New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




