Applying LC and LC–MS Methods to the Characterization and Analysis of rAAV
A look at the LC and LC–MS methods that are helping to overcome challenges in sample analysis, and how this can make adeno‑associated virus (AAV)–based gene therapy more accessible.
Because of advances in the fundamental understanding of gene therapy treatments and the 2017 regulatory approval of a recombinant adeno-associated viral (rAAV) therapy, interest in developing rAAV-based therapies has increased. However, for patients worldwide to truly benefit from these therapies, significant improvements in the efficacy and manufacturing efficiency of rAAV are essential. We have recently seen substantial increases of inquiries regarding liquid chromatography (LC) and liquid chromatography–mass spectrometry (LC–MS) methods for the characterization and analysis of rAAV from scientists who are supporting advancements in product development. This edition of “Column Watch” highlights selected LC and LC–MS methods that can be applied to the analysis of these complex products.
Gene therapy involves altering of the genome of selected cells within a patient (1). Although CRISPR/Cas9 has received widespread attention because of the groundbreaking work done by Emmanuelle Charpentier and Jennifer A. Doudna that resulted in their receiving the 2020 Nobel Prize in Chemistry, there are a number of other gene therapy methodologies currently being studied and developed (1).
One such approach is the in vivo delivery of a genetic payload to specific patient cells through the use of a non-replicating virus (viral vector). In this approach, the primary functions of the virus capsid are to shield the encapsulated RNA or DNA from enzymatic degradation and to deliver it to the interior of the cell. The predominant virus types adopted for therapeutic development are recombinant adeno-associated virus (rAAV), lentivirus, as well as adenovirus. In this article, we focus on liquid chromatography (LC) and liquid chromatography–mass spectrometry (LC–MS) characterization and analysis of rAAV, which continues to be the most widely used viral gene therapy platform (2). This focused perspective is predicated on the wide use of rAAV and our direct experience with rAAV samples.
Before considering analytical approaches, it is not only useful to study the nature of rAAV structure, but to also understand the general manufacturing process. A rAAV consists of a ~25 nm diameter icosahedral protein shell or capsid that is composed of three different viral proteins (VP1, VP2, and VP3) arranged in pentameric substructures and in its entirety each capsid contains 60 VP proteins in a 1:1:10 proportion of VP1:VP2:VP3 (3). The VP proteins are highly homologous, where VP2 (~65 kDa) is composed of the entire amino acid sequence of VP3 (~60 kDa) with an N‑terminal extension, and VP1 (~80 kDa) is an N‑terminal extension of VP2. The entire rAAV protein capsid shell has a molecular weight of approximately 3.7 MDa. Although also homologous, the varied serotypes of AAV are defined by variation in their capsid protein primary structures. It is these differences that can dictate viral tropism resulting in a gene therapy more selectively targeting the desired cells of a patient. The single‑stranded DNA (ssDNA) of therapeutic rAAV is located in the interior of the capsid structure and is composed of only the therapeutic gene and sequence regions that are required for the expression of the protein encoded by the therapeutic gene. Because of its size, the natural ssDNA packaging limit of rAAV is restricted to 4.7 kilobases (kb) with a molecular weight of approximately 1.55 MDa and extending that limit has been shown to be problematic (4). With the protein and ssDNA the total molecular weight of rAAV is approximately 4.3 MDa.
The rAAV production process can be quite varied and is a highly active area of study. A common production method is to transiently transfect a suspended host cell culture with plasmids that, when incorporated into the host cell, are designed to result in the host cell producing the rAAV capsid containing the desired therapeutic gene as ssDNA. In this approach, the plasmids used for the transient transfection may be considered critical reagents in the manufacturing process. As a result, these plasmid preparations may need to be well characterized and controlled (4). The harvesting and purification of rAAV generally includes cell lysis, clarification, and purification. Purification steps include a variety of extraction, immunoaffinity, chromatographic, and filtration steps (5). The influences that such a broad range of cell culture and purification steps can have on therapeutic rAAV quality are an important consideration when developing a structural characterization and product testing strategy.
In addition to the complexity of the rAAV structure, there are several additional challenges that are presented by rAAV samples compared to typical recombinant protein analyses. The low productivity of the rAAV manufacturing process results in limited sample quantities, and although this may not be a major issue for the final commercialized product, it can be very problematic when assessing process development samples where often only microgram quantities of protein and ssDNA components of rAAV are available to the analyst. Another challenge of rAAV samples is their low concentrations. As examples, a formerly approved intramuscular rAAV therapy was formulated at a concentration of 3 × 1012 vector genomes per millilitre (vg/mL) and the concentration of a current ocular rAAV therapy is 5 × 1011 vg/mL. This translates into protein concentrations of 20 μg/mL and 3 μg/mL, and ssDNA concentrations (assuming 4.7 kb ssDNA) of 13 μg/mL and 2 μg/mL, respectively. To add further complication, VP1 and VP2 each account for 8% of the total number of proteins, resulting in single to submicrogram levels of these two proteins in the formulated samples.
Although an in-depth review on all of the methods that may be included as part of rAAV product characterization and analysis is well beyond the scope of this article, we highlight several LC and LC–MS methods that we have applied to characterize rAAV, as well as some related studies. Included in this column are the anion‑exchange chromatography (AEC) and size‑exclusion chromatography (SEC) analyses of intact virus, the LC–MS analysis of intact VP proteins, and peptide mapping of VP proteins.
Anion-Exchange Chromatography for Intact Capsid Analysis
During the rAAV biomanufacturing process, empty capsids that do not contain the desired ssDNA are also produced. Without a consistent ssDNA payload, the efficacy and safety of rAAV therapies may be compromised (6). Several techniques have been used to quantify empty capsids in AAV samples, including analytical ultracentrifugation (AUC), static spectrophotometry, cryogenic electron microscopy (Cryo-EM), and charge detection MS (CDMS) (7–10). However, various challenges are presented by these methods including the cost of instrumentation, the expertise needed, the sample amounts required, low sample throughput, and susceptibility to interferences. As a result, the development of analytical methods to measure empty capsid levels remains an active area of research.
AEC was reported to have the potential of separating empty and full capsids based on differences in surface charge (9,11,12). It was also shown that compared to UV absorbance detection, fluorescence detection not only increases signal‑to‑noise ratio, but also has much less response bias between empty and full capsids, which is in part due to minimal intrinsic fluorescence of DNA (2). Figure 1 shows the optimized separation of AAV8 empty and full capsids using 5 µm non-porous strong anion-exchange particles with a quaternary ammonium ligand. It was also observed (data not shown) that AEC retention varied by rAAV serotype indicating that optimized method conditions are serotype dependent. In addition, the percentage of empty capsids in a sample can be determined once the relative intrinsic protein fluorescence response between the empty and full capsids is determined.


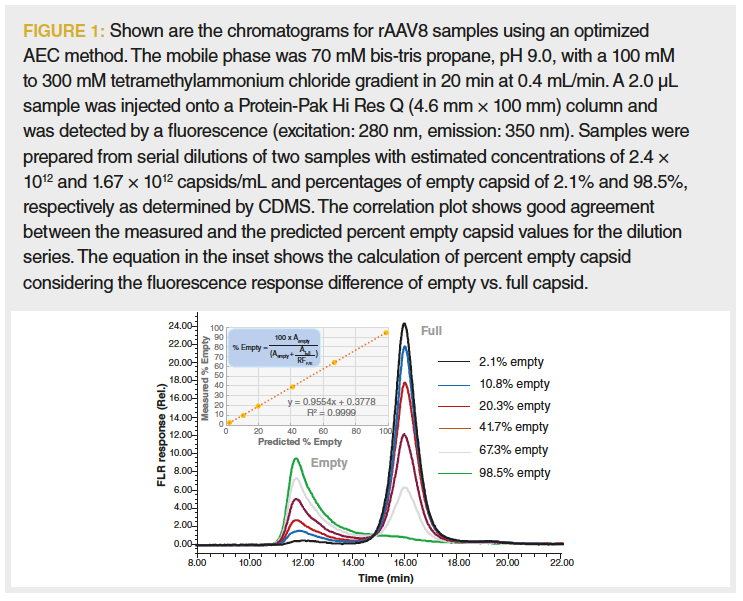
In the chromatographic overlay, we also observe that for the 98.5% empty sample (as confirmed by CDMS, data not shown), a substantial portion of the empty capsids were coeluted with the full capsid fraction (13). This is presumed to be because of the charge heterogeneity of the empty capsid proteins or the presence of oligonucleotide fragments on the empty capsid surface (14). These results indicate that an accurate assessment of partially full AAV8 capsid may not be possible to obtain via AEC. Despite this potential limitation, AEC may provide a useful approach to guide the process and product development and monitor product quality because of its simplicity, low sample demand, and precision.
SEC for Aggregate, Fragment, and Concentration Analysis
The levels of virus aggregation may be considered to be a critical quality attribute (CQA) in therapeutic rAAV preparations (4,15). SEC is most often employed for the analysis of aggregation and fragmentation of proteins such as IgG, due in large part to the precision and sample throughput of the analysis. There are limited reports of its use for the analysis of rAAV; in contrast, the routine use of dynamic light scattering (DLS) has been more reported (4,16,17). It is important to recognize that the upper analyte size limitation for SEC separations is approximately 100 to 200 nm in diameter, which is in the subvisible range (18). Above these limits, resolution will be low, and the analyte may be altered because of on-column shear forces, or trapped by the column. Therefore, these subvisible entities in rAAV preparations are analyzed using DLS, nanoparticle tracking analysis (NTA), and asymmetric flow field-flow fractionation (AF4) methods, among others (18,19).
As previously noted, rAAV has a total protein and ssDNA molecular weight of 4.3 MDa. However, the compact icosahedral structure has a diameter of only 25 nm (20). By comparison, IgG protein has a reported maximum lateral dimension of approximately 15 nm and its dimer and lower valency high molecular weight forms (HMW) are well resolved on a column packed with 200 Å pore‑size diol‑bonded silica‑hybrid SEC particles (21). Therefore, it was predicted that a high efficiency 450 Å pore-size version of that column should provide the required separation of rAAV HMW forms ranging in valency from dimer to tetramer. In addition to an increase in precision, SEC analysis can also provide information on the capsid fragments and other proteins in rAAV preparations in comparison to other methods.
Figure 2 shows the optimized non‑denaturing SEC separations for several rAAV-CMV-GFP serotypes containing ssDNA with CMV as the promoter sequence and green fluorescent protein (GFP) as the insert gene (22). To accommodate the low concentration of the rAAV samples, a detector monitoring intrinsic protein fluorescence was deployed (23). Fluorescence provides approximately 10-fold greater signal-to-noise in comparison to UV absorbance at 280 nm or 260 nm. Alternatively, the addition of a multiangle light scattering (MALS) detector to the SEC analysis of rAAV has also been shown to provide enhanced sensitivity for virus aggregates and in addition can provide accurate information on the capsid concentration and an estimate of empty capsid levels (17). Rapid estimates of these two sample characteristics can also be derived from an SEC separation using an UV absorbance detector or a fluorescence and UV absorbance detector in series (24,25).



LC–MS for Intact VP Protein Analysis
Interrogation of proteins that compose the capsid of rAAV is an integral aspect of rAAV characterization. At a most basic level, the 1:1:10 proportion of VP1:VP2:VP3 proteins that comprise the capsid correlate with the effectiveness of an rAAV preparation (26). Changes in this proportion are not only observed for different cell culture systems but can also be a measure of process consistency. Traditionally, sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) has been used to evaluate the relative VP protein abundance and more recently the use of SDS–capillary gel electrophoresis (SDS-CGE) has been adopted. In contrast, LC–MS separations of VP capsid proteins can not only confirm consistent VP1:VP2:VP3 ratios but can also provide useful information on VP degradation (such as deamidation and oxidation), post-translational modifications (PTMs) such as phosphorylation and SUMOylation, and engineered point mutations that may impact the efficacy of rAAV products (26,27). For rAAV subserotypes with engineered point mutations, LC–MS analysis of the VP proteins may also find utility as a subserotype identity test.
The most commonly used LC–MS separation method for the analysis of proteins is reversed-phase LC (RPLC), which has been effectively applied to the analysis of rAAV (28). Building on this work, a method was developed for the RPLC-MS analysis of several rAAV serotypes as shown in Figure 3 (29). The separation was optimized for a set of rAAV serotypes using a 300 Å pore size C4 column and difluoroacetic acid (DFA) as a mobile‑phase modifier and analyzed using a compact time‑of‑flight LC–MS system (LC–TOF‑MS). When deployed, fluorescence optical detection was favoured over UV absorbance due to the nearly 50‑fold greater sensitivity. Figure 3 also shows representative mass spectra for the major components observed, including significant levels of phosphorylation of the VP1 and VP2 proteins.



A well-executed alternative approach to LC–MS analysis of the VP proteins using a hydrophilic interaction liquid chromatography (HILIC) separation with MS detection has also been reported (30). This separation makes use of a 300 Å pore size, silica‑hybrid based amide HILIC column that was optimized for the separation of glycosylated peptides and proteins (31,32). The reported HILIC separation can be applied to a wide range of rAAV serotypes, and although trifluoroacetic acid (TFA) is used as a mobile‑phase modifier, MS sensitivity is enhanced by modifying the MS desolvation gas with propionic acid and isopropanol.
LC–MS for VP Protein Peptide Mapping
As discussed for intact VP proteins, the LC–MS identification and analysis of PTMs and protein degradation can provide useful information to both guide rAAV product development and monitor product quality (27). However, to evaluate site‑specific variants, peptide mapping may likely be required. Additionally, a peptide mapping procedure can also be deployed to monitor for host cell proteins (HCP) that may be of concern in rAAV products (33). A peptide mapping procedure involves the proteolytic digest of a protein and may also include disulfide bond reduction and alkylation steps. The LC or LC–MS analysis of the digested sample can be comprehensive to cover as much of the protein amino acid sequence as possible or be focused on the peptides and peptide variants of interest.
In comparison to typical recombinant protein therapeutics, rAAV samples present several significant challenges during peptide map development. As noted previously, these samples are much lower in concentration in addition to presenting the difficulties presented by the homology and relative abundance of the VP proteins. Furthermore, the amount of sample that can be used for these analyses is often limited and rAAV samples are often formulated with surfactants that can significantly degrade rAAV MS signal quality. Another choice that can be made for this analysis is whether to map the capsid proteins as a single digest or to purify VP1, VP2, and VP3 prior to digestion.
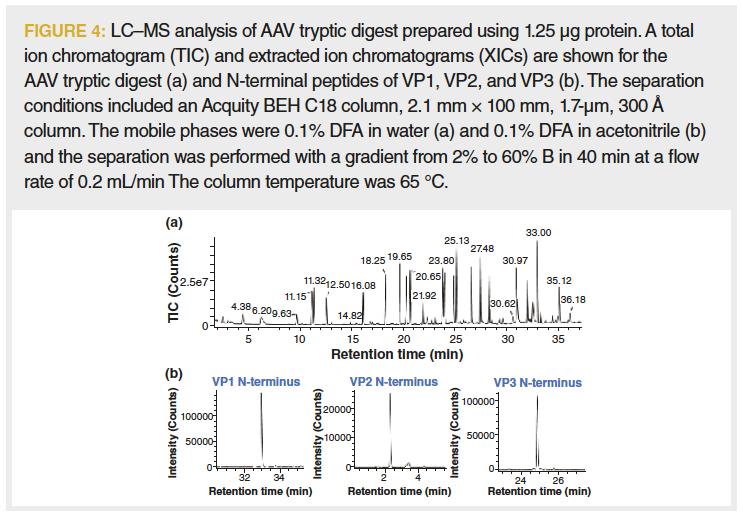
Given the limited amounts of sample that are often available, a single‑digest mapping method was developed. The disadvantage of this approach is that variants in the conserved sequence domains cannot be attributed to a VP protein type. The use of a single digest also presents a dynamic range challenge for UV absorbance detection of the peptide map given the greater than 10‑fold abundance of VP3 domain peptides vs. the peptides that comprise the N-terminal extensions of VP2 and VP1. With these considerations, a tryptic digest of 1.25 μg of rAAV was optimized and then analyzed using an LC–TOF‑MS system without a UV absorbance detector and resulted in sequence coverages of greater than 95% for VP1, VP2, and VP3 (Figure 4).


Conclusions
As the clinical interest in rAAV gene therapies increases it will be critical to optimize the product efficacy and the efficiency of the rAAV manufacturing process. LC and LC–MS technologies have a long history of use in the development, characterization, and quality testing of recombinant protein therapies and will likely be valuable tools in the advancement of rAAV‑based gene therapies, as well as other viral and nonviral gene therapy development. Indeed, as fairly recently‑introduced therapy modalities, the regulatory analytical requirements for these products are only starting to mature (34). This article has provided a brief introduction to the potential application of some of these LC and LC–MS methods to rAAV characterization and analysis. In addition, we have also seen that in comparison to recombinant protein analysis, the deployment of LC and LC–MS methods is further complicated by numerous factors such as more complex samples and limited sample amounts.
Acknowledgements
The authors would like to thank Benjamin E. Draper of Megadalton Solutions for providing CDMS results for the rAAV8 samples used for AEC method development.
References
- C.E. Dunbar, K.A. High, J.K. Joung, D.B. Kohn, K. Ozawa, and M. Sadelain, Science 359(6372), eaan4672 (2018).
- C. Wang, S.H.R. Mulagapati, Z. Chen, J. Du, X. Zhao, G. Xi, L. Chen, T. Linke, C. Gao, A.E. Schmelzer, and D. Liu, Mol. Ther. 15, 257–263 (2019).
- Y.Z. Tan, S. Aiyer, M. Mietzsch, J.A. Hull, R. McKenna, J. Grieger, et al., Nat. Commun. 9, 362–368 (2018).
- J.F. Wright, Gene Therapy 15, 840–848 (2008).
- D. Blessing, N. Deglon, and B.L. Schneider, in Scalable Production and Purification of Adeno-Associated Viral Vectors (AAV) (Springer, New York, 2018), pp. 259–274.
- K. Gao, M. Li, L. Zhong, Q. Su, J. Li, S. Li, R. He, Y. Zhang, G.Hendricks, J. Wang, and G. Gao, Mol. Ther. Methods. Clin. Dev. 1(9), 20139 (2014).
- B. Burnham, S. Nass, E. Kong, M. Mattingly, D. Woodcock, A. Song, S. Wadsworth, S.H. Cheng, A. Scaria, and C.R. O’Riordan, Hum. Gene Ther. Methods 26(6), 228–242 (2015).
- J.M. Sommer, P.H. Smith, S. Parthasarathy, J. Isaacs, S. Vijay, J. Kieran, S.K. Powell, A. McClelland, and J.F. Wright, Mol. Ther. 7(1), 122–128 (2003).
- S. Subramanian, A.C. Maurer, C.M. Bator, A.M. Makhov, J.F. Conway, K.B. Turner, J.H. Marden, L.H. Vandenberghe, and S.L. Hafenstein, Hum. Gene Ther. Methods 30(12), 1449–1460 (2019).
- E.E. Pierson, D.Z. Keifer, A. Asokan, and M.F. Jarrold, Anal. Chem. 88(13), 6718–6725 (2016).
- E.D. Horowitz, K.S. Rahman, B.D. Bower, D.J. Dismuke, M.R. Falvo, J.D. Griffith, et al., J. Virol. 87, 2994–3002 (2013).
- X. Fu, W.C. Chen, C. Argento, P. Clarner, V. Bhatt, R. Dickerson, et al. Hum. Gene Ther. Methods. 30, 144–152 (2019).
- H. Yang, S. Koza, and W. Chen, Anion-exchange chromatography for determining empty and full capsid contents in adeno-associated virus 2020. Application Note (2020).
- G. Qu, J. Bahr-Davidson, J. Prado, A. Tai, F. Cataniag, J. McDonnell, et al. J. Gen. Mol. Virol. 140, 183–192 (2007).
- T. Tanaka, H. Hanaoka, and S. Sakurai, Eur. J. Pharm. 155, 88–102 (2020).
- Y. Yang, Z. Su, G. Ma, and S. Zhang, Eng. Life Sci. 20(11), 451–465 (2020).
- M. Chen and A. Purchel, AAV critical quality attribute analysis by SEC-MALS. Application Note 1617. wyattcom.
- J.F. Carpenter, T.W. Randolph, W. Jiskoot, D.J. Crommelin, C.R. Middaugh, and G. Winter, et al. J. Pharm. Sci. 98(4), 1201–1205 (2009).
- B. Slutter and W. Jiskoot, Expert Opin. Drug Deliv. 13(2), 167–170 (2016).
- S.K. Vernon, J.T. Stasny, A.R. Neurath, and B.A. Rubin, J. Gen. Virol. 10, 267–272 (1971).
- L.J. Harris, E. Skaletsky, and A. McPherson, J. Mol. Biol. 275, 861–872 (1998).
- W. Xiong, A.E. MacColl Garfinkel, Y. Li, L.I. Benowitz, and C.L. Cepko, J. Clin. Investig.125, 1433–1445 (2015).
- S. Koza and W. Chen, Size-Exclusion Chromatography Analysis of Adeno-Associated Virus (AAV) Preparations Using a 450 Å Diol-Bonded BEH Column and Fluorescence Detection 2020. Application Note (2020).
- S. Koza and W. Chen, Rapid AAV Concentration Determination Using Size-Exclusion Chromatography with Fluorescence and UV Dual Detection 2020. Application Note (2020).
- H. Runnels, ed. Gene Therapy and AAV: Advancing Analytical Characterization and Product Understanding for Late-Stage Success (WCBP, Washington DC, 2019).
- S. Pacouret, M. Bouzelha, R. Shelke, E. Andres-Mateos, R. Xiao, A. Maurer, et al. Mol. Ther. 25, 1375–1386 (2017).
- B. Mary, S. Maurya, S. Arumugam, V. Kumar, and G.R. Jayandharan, The FEBS Journal 286, 4964–4981 (2019).
- X. Jin, L. Liu, S. Nass, C. O’Riordan, E. Pastor, and X.K. Zhang, Hum. Gene Ther. Methods28(5), 255–267 (2017).
- X. Zhang, S.M. Koza, Y.Q. Yu, and W. Chen, Optimizing Adeno-Associated Virus (AAV) Capsid Protein Analysis Using UPLC and UPLC-MS. Waters Application Note. 2020, EN72000686.
- A.P. Liu, S.K. Patel, T. Xing, Y. Yan, S. Wang, and N. Li, J. Pharm. Biomed. 189, 113–481 (2020).
- M.A. Lauber, S.M. Koza, P.C. Iraneta, and K.D. Wyndham, Materials for hydrophilic interaction chromatography and processes for preparation and use thereof for analysis of glycoproteins and glycopeptides. Google Patents, 2015.
- A. Periat, S. Fekete, A. Cusuman, J.L. Veuthey, A. Beck, M. Lauber, et al. J. Chromatogr. A.1448, 81–92 (2016).
- J.F. Wright, Biomedicines 2(1), 80–97 (2014).
- J. Alcudia and J.F. Wright, JBT31, s43 (2020).
Authors
Ximo Zhang is a Senior Scientist at Waters Corporation in Milford, Massachusetts, USA.
Hua Yang is a Principal Scientist at Waters Corporation in Milford, Massachusetts, USA.
Steve Koza is a Consulting Scientist at Waters Corporation in Milford, Massachusetts, USA.
Weibin Chen is Director, Scientific Operations at Waters Corporation in Milford, Massachusetts, USA.
Column Editor
David S. Bell is a director of Research and Development at Restek. He also serves on the Editorial Advisory Board for LCGC and is the Editor for “Column Watch.” Over the past 20 years, he has worked directly in the chromatography industry, focusing his efforts on the design, development, and application of chromatographic stationary phases to advance gas chromatography, liquid chromatography, and related hyphenated techniques. His main objectives have been to create and promote novel separation technologies and to conduct research on molecular interactions that contribute to retention and selectivity in an array of chromatographic processes. His research results have been presented in symposia worldwide, and have resulted in numerous peer-reviewed journal and trade magazine articles.
Direct correspondence to: amatheson@mjhlifesciences.com










New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.





