Structured Approach to Method Development for Bioanalytical HILIC–MS-MS Applications
Special Issues
A structured, general purpose approach to method development for bioanalytical hydrophilic interaction chromatography–tandem mass spectrometry (HILIC–MS-MS) applications is described.
Hydrophilic interaction chromatography (HILIC) is a unique mode of chromatography in which polar compounds are well retained and resolved using mobile phases containing high concentrations of organic solvent. Because many drug metabolites are polar, quantification using reversed-phase liquid chromatography (LC) can be compromised by poor retention and the resulting ion suppression with mass spectrometry (MS) detection. HILIC provides good retention of polar metabolites outside the suppression region and thus allows sensitive and accurate quantification. This combination of retention of polar compounds and high organic in the mobile phase makes this mode of chromatography uniquely suited to high sensitivity, quantitative bioanalytical LC–MS-MS applications.
Despite these advantages, the use of HILIC mode separations has grown relatively slowly since its inception over a decade ago. Several possible explanations for the slow acceptance and implementation of HILIC methods include the perception that HILIC methods lack robustness. This misperception may stem from the use of conditions common in reversed-phase LC with HILIC methods. For example, the use of an acid such as formic acid to control pH is common in reversed-phase LC but in general does not provide good performance in the majority of HILIC applications (1). Another possible contributing factor may be inattention to the pronounced effect high acetonitrile concentration has on buffer and analyte pKa. This is also commonly overlooked in reversed-phase LC methods but must be considered when using HILIC. When HILIC conditions are chosen carefully, robust methods can be routinely obtained. To provide a framework for HILIC users, a rationally designed, structured approach to HILIC method development is described. The conditions proposed have shown good general purpose performance with a wide variety of compound classes. This approach is specifically targeted for optimizing bioanalytical applications by evaluating analyte selectivity, overall separation performance, and MS signal suppression simultaneously using a variety of robust separation conditions. Optimum conditions can then be chosen based on both separation performance and signal suppression.
Experimental
Apparatus
The chromatographic system consisted of an Agilent 1100 series binary pump (Palo Alto, California), on-line solvent degasser, autosampler, and an Applied Biosystems API3000 tandem mass spectrometer with TurboIonSpray ESI interface (Foster City, California). The chromatographic system was controlled using Analyst 1.41 software.
Chromatographic separations were performed on a 100 mm × 2.0 mm, 3-μm Phenomenex Luna HILIC, column and a 100 mm × 2.0 mm, 3-μm Phenomenex Luna Si (2) column (Torrance, California).
Chemicals
Heroin, morphine, normorphine, morphine-3-B-glucuronide, codeine, and norcodeine (1 mg/mL in methanol) were obtained from Cerilliant (Round Rock, Texas). Acetonitrile was obtained from Honeywell Burdick and Jackson (Muskegon, Michigan), formic acid from EMD (Gibbstown, New Jersey), and ammonium hydroxide from Fisher Scientific (Pittsburgh, Pennsylvania). Ammonium acetate and ammonium formate were obtained from Sigma Aldrich (St. Louis, Missouri). The ammonium acetate buffer was prepared by dissolving 7.7 g ammonium acetate salt in 1 L of Milli-Q water. Acetic acid (5.4 mL) was added to obtain a pH of 5.8. The ammonium formate buffer was prepared by dissolving 6.3 g ammonium formate salt in 1 L of Milli-Q water. Formic acid (10.4 mL) was added to obtain a pH of 3.2. Neat standards (1000 ng/mL each) were prepared by adding 20 μL of each 1 mg/mL stock solution to 19.9 mL 90:10 acetonitrile–100 mM ammonium formate (pH 3.2). Human plasma disodium EDTA was obtained from Valley Biomedical (Winchester, Virginia).
Sample Preparation
Protein precipitation was performed using the following procedure. A 1-mL volume of human plasma was added to 5 mL acetonitrile, vortexed, then centrifuged at 10,000 rpm for 5 min. The supernatant was removed and analyzed without further treatment.
Chromatographic Conditions
All separations were performed using the conditions shown in Table I. The initial and final gradient mobile phase compositions were premixed and placed on the "B Channel" and "A Channel" of the binary pump, respectively. Channel B was the high organic or initial mobile phase and consisted of a mixture containing 90:5:5 (v/v/v) acetonitrile, water, and buffer, where buffer is 100 mM ammonium acetate or formate. Channel A was the low organic or final mobile phase and consisted of a mixture containing 50:45:5 (v/v/v) acetonitrile, water, and buffer, where buffer is 100 mM ammonium acetate or formate. All analytes were detected using tandem MS in positive ion mode with the mass transitions and source conditions shown in Table I.



Table I: Chromatographic and detector conditions
Results and Discussion
Method Development Overview
The primary chromatographic parameters available to adjust retention and selectivity in HILIC are sorbent chemistry, type and concentration of organic modifier, pH, ionic strength, and temperature. The first three are generally the most influential, and therefore only these are varied during method development. The other parameters are generally less influential and can be varied during optimization. The method development strategy employed was to quickly determine if an analyte is a good candidate for HILIC and to get a good approximation of the best separation conditions by using a rationally chosen set of conditions that provide the most information in the least time. These experiments also provide the basis for further optimization. The proposed method development approach utilizes an acetonitrile gradient with three different column chemistries at two buffer pH values. All separations are performed at room temperature. Gradient elution is used because it is the most efficient starting point for method development. The 100 mm × 2.0 mm column format was chosen because it provides good resolving power at flow rates compatible with most MS sources. The buffer pH values chosen span the pKa range of many weak acids and bases and also are available with MS-compatible buffers. Evaluating selectivity and MS suppression using this variety of conditions allows selection of the best overall analysis conditions.
Three opiate drugs and their metabolites were chosen as probe compounds to illustrate the method development approach. These compounds contain a variety of functionalities as well as a wide range of pKa (7 units) and log P values (2.5 units). The probe compounds are shown in Figure 1 and illustrate the similarities in structure. The primary differences among the majority of the compounds are the addition or subtraction of one or two methyl or acyl groups. The glucuronide metabolite of morphine is the most chemically dissimilar compound, containing a carboxylic acid and a sugar group. Removal of a methyl or acyl group produces an aromatic hydroxy or converts a tertiary amine to a secondary. These changes affect the pKa, hydrogen bonding, and hydrophobicity of the compounds, which will affect their retention and selectivity.


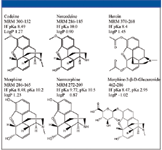
Figure 1: Structure, MRM transitions, and physical properties of the basic test probes heroin, morphine, normorphine, morphine-3-B-glucuronide, codeine, and norcodeine.
Evaluation of Chromatographic Performance
As shown in Table I, an inverse acetonitrile gradient is used since acetonitrile is the weak solvent and water is the strong solvent in HILIC. The gradient profile includes a 1.7-min isocratic hold at 90% acetonitrile to determine if a compound is weakly retained in HILIC mode. A final 1.7-min isocratic hold at 50% acetonitrile is included to determine if a compound is strongly retained. The gradient portion of the elution profile is the ideal elution region. A compound eluting during the gradient will allow the maximum flexibility in adjusting selectivity. A few general recommendations regarding gradient elution in HILIC mode: three injections per column per set of conditions should be made to test reproducibility and column equilibration. Column equilibration typically requires at least 10 column volumes at 90% acetonitrile, but this varies significantly with column chemistry. Bare silica, for example, requires 15–20 column volumes for equilibration while Luna HILIC requires ~10 column volumes. Complete equilibration generally is not required for routine methods but is required in method development to allow estimation of appropriate isocratic conditions.
The second parameter varied is column chemistry. The three sorbent chemistries tested were Luna 3-μm HILIC, Luna 3-μm Si (2), and SeQuant ZIC-HILIC 3-μm (zwitterion). These sorbents were chosen because they have been shown to give the largest differences in selectivity. As shown in Figure 2, the Luna HILIC column gives good resolution and peak shape for all compounds at pH 5.8. All compounds are resolved on the zwitterion column but the peak shapes are generally a bit poorer. On the silica column, peaks 2 and 4 and peaks 3 and 5 are coeluted. Analyte coelution can cause ion suppression so it should be avoided. The elution orders between the Luna HILIC and zwitterion columns are the same although the peak spacing is different. On the Luna Si column, the elution order of peaks 3 and 4 is reversed from the other columns. The resolution value for a pair of closely eluted peaks that share a transition is also shown in the figure. Because these compounds share a transition, they must be resolved chromatographically. These chromatograms illustrate the important role sorbent chemistry plays with selectivity in HILIC.


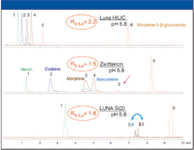
Figure 2: Effect of sorbent chemistry on retention and selectivity.
The third parameter varied in method development is buffer pH. The recommended pH values of 3.2 and 5.8 are aqueous pH values. The mobile phase pH is quite different from the aqueous pH due to the effect of organic solvent on buffer component pKa. In general, the pKa of weak acids increases with increasing organic concentration and the pKa of weak bases decreases with increasing organic concentration (2). Familiarity with these trends is typically sufficient to understand pH effects in HILIC. As shown in Figure 3, pH has a significant effect on the retention and selectivity of many compounds. With the Luna HILIC sorbent, peaks 3 and 4 are resolved with an aqueous buffer pH of 5.8 but are coeluted at pH 3.2. Also, the retention for some compounds decreases while others increase at pH 3.2. With the Luna Si (2) sorbent, significant selectivity changes are observed with pH changes. At pH 3.2, peaks 2 and 4 are resolved and their retention times shifted in opposite directions. These examples demonstrate that buffer pH is an important tool for manipulating the retention and selectivity of ionizable compounds in HILIC.


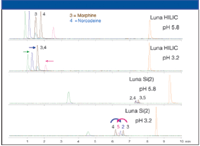
Figure 3: Effect of pH on selectivity and retention.
Once the three primary parameters (sorbent chemistry, percent organic, and buffer pH) have been explored, the selectivity results can be evaluated. The best results obtained with each column are compared and, as shown in Figure 4, all columns give acceptable chromatographic performance. The next consideration in method development is matrix effects.


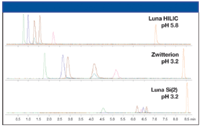
Figure 4: Optimum chromatographic performance obtained with each sorbent.
Evaluation of Matrix Effects
The experiments to assess matrix effects can be run along with the selectivity experiments to maximize productivity. This allows evaluation of matrix effects at different pH values on each sorbent. The approach used is similar to that described by others (3). A syringe pump is used to infuse a standard solution containing all analytes of interest into the column effluent through a tee just before the MS source. This infusion experiment sets up a high, stable background signal on all analyte transitions. An extracted blank sample of protein-precipitated plasma is then injected and eluted from the column. Any changes in the analyte signals during elution indicate possible matrix effects. This approach allows for a qualititative, visual assessment of matrix effects. The profiles for the three columns tested at pH 3.2 are shown in Figure 5, and significant column-to-column differences are apparent. This result further reinforces the importance of testing several sorbents during bioanalytical method development and that matrix effects are highly compound specific. Any changes in analyte signal coming from changes in mobile phase composition are reproducible and will not affect quantitative performance. While not shown here, mobile phase pH can also have a major effect on ion suppression and should be evaluated. A useful way to combine the selectivity and suppression data is to overlay the selectivity standard and the suppression chromatograms at the same pH. The Luna HILIC column showed its best selectivity at pH 5.8, so these chromatograms are overlaid. As shown in Figure 6, the suppression early in the chromatogram doesn't directly interfere with any analyte but some robustness testing should be performed to ensure that analyte retention times and the suppression region do not change with minor changes in conditions. No significant matrix effects are apparent for the other analytes since there is no overlap between suppression or enhancement with the retention time of the analytes. This testing is designed to give a qualitative overall picture of matrix effects and to determine if the analytes are eluted near or within affected regions. After promising conditions have been identified, further quantitative testing of matrix effects should be performed.


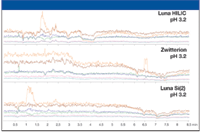
Figure 5: MS matrix effects profiles for each chromatographic sorbent with protein precipitated blank human plasma.
Conclusions
This research shows the utility of a rationally designed, structured approach to HILIC method development. The conditions outlined have shown good general purpose performance with a wide variety of compound classes. This approach is specifically targeted for optimizing bioanalytical applications by evaluating analyte selectivity, overall separation performance, and MS signal suppression simultaneously using a variety of robust separation conditions. Optimum overall conditions can then be chosen based on both separation performance and matrix effects.


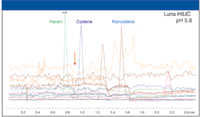
Figure 6: Comparison of chromatographic performance and matrix effects.
A. Carl Sanchez and Monika Kansal are with Phenomenex, Inc., Torrance, California.
References
(1) A.C. Sanchez and M. Kansal, LCGC The Peak, 16–24 (November 2007).
(2) M. Roses and E. Bosch, J. Chromatogr., A 982, 1–30 (2002).
(3) R. King, R. Bonfiglio, C. Fernandez-Metzler, C. Miller-Stein, and T. Olah, J. Am. Soc. Mass. Spectrom. 11, 942–950 (2000).













Assessing Thorium-Peptide Interactions Using Hydrophilic Interaction Liquid Chromatography
February 4th 2025Paris-Saclay University scientists used hydrophilic interaction liquid chromatography (HILIC) coupled to electrospray ionization mass spectrometry (ESI-MS) and inductively coupled plasma mass spectrometry (ICP-MS) to assess thorium’s interaction with peptides.



