Stir Bar Sorptive Extraction (SBSE) and Headspace Sorptive Extraction (HSSE): An Overview
LCGC North America


The guest columnists review SBSE and HSSE and examine their advantages and limitations, along with providing approaches to overcoming the latter.
The increase in the number of controls an analytical laboratory must make that has occurred over the last 15–20 years has induced a radical change in the approach to chemical analysis. The present trend for routine laboratories, in particular those where large numbers of homogeneous samples must be analyzed, is to develop, wherever possible, the so-called "Total Analysis Systems" (TAS) — that is, automatic systems in which sample preparation and analysis are merged into a single step, with the aim of reducing the workload to a minimum. This need has influenced greatly the development of both the main steps of an analytical procedure, that is, sample preparation and analysis, and within sample preparation it has increased greatly the importance of solvent-free sample preparation techniques (that is, techniques whereby an analyte or analytes are isolated from a matrix without using a liquid solvent).



Ronald E. Majors
Stir-bar sorptive extraction (SBSE) belongs to a group of techniques that have been developed in view of the previously mentioned analytical approach. This article is an overview on both SBSE and headspace sorptive extraction (HSSE) about 10 years after their introduction mainly focusing on their evolution after David and colleagues' review (1) published by LCGC in 2003 and on their perspectives of development.
The Technique
SBSE was introduced in 1999 by Pat Sandra's group to overcome some of the limits of the existing techniques, in particular in the recovery of medium-to-high volatility analytes when sampled in liquid phase with polydimethylsiloxane-open tubular traps (PDMS-OTT); a further aim was to improve the limited recovery achievable in ultratrace analysis with solid-phase microextraction (SPME), especially under unfavorable phase ratios when working with small volumes of sorptive material (in general PDMS) coating the fused-silica fiber (2). SBSE was first developed for sampling in liquid phase and is based upon sorption of the investigated analytes or fraction onto a very thick film of PDMS coated onto a glass-coated magnetic stir bar (commercially known as Twister, Gerstel GmbH, Muelheim, Germany). Figure 1a gives a scheme of a conventional PDMS SBSE device. Sampling is done by directly introducing the SBSE device into the aqueous sample; in the original experiments, the analytes sampled for a given time were recovered by thermal desorption and then on-line transferred to a gas chromatography (GC) or GC–mass spectrometry (MS) system for analysis. Later, liquid desorption in combination with high performance liquid chromatography (HPLC) also was applied, mainly for analytes not analyzable by GC (3,4).

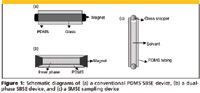
Figure 1
Basic Concepts of SBSE
SBSE is based upon sorption, which is a form of partition based upon the analyte's dissolution in a liquid-retaining polymer from a liquid or vapor sample, thus, originating a bulk retention (5–9). The main advantages of sorption are related to high inertness of PDMS, which gives better performance for labile, polar, or reactive compounds; absence of catalytic degradation reactions; analyte recovery mechanism based upon a well-known chromatographic process; and linearity of sorption isotherms, which is fundamental for quantitative analysis.
Sandra's group also advanced a theory for SBSE that was rather similar to that of SPME. It was based upon the approximation that the partitioning coefficient of an analyte between PDMS and water is proportional to its octanol–water partition coefficient (KO/W); thus, its theoretical recovery can be calculated through the following equation:
Rth = mPDMS/m0 = α/1 + α
where α = KO/W/β and + mPDMS is the mass of the analyte present in the PDMS, m0 is the total amount of analyte originally present in the water sample, and β is the volume of water/volume of PDMS phase ratio.
The use of SBSE was extended almost immediately to sampling in vapor phase (headspace) by Bicchi and colleagues (5) and Tienpont and colleagues (6); the technique is known as HSSE. In HSSE, sampling is operated in static mode by suspending the PDMS SBSE device in the vapor phase in equilibrium (or not) with the solid or liquid matrix. Again, analyte recovery is by thermal desorption combined on-line with GC or GC–MS analysis of the desorbed analyte.
SBSE and HSSE have been the object of many reviews, singly or jointly or within surveys on sample preparation of specific classes of compounds or matrices (8–21).
SBSE Advantages and Limits
SBSE and HSSE immediately enjoyed considerable success, and numerous applications in different fields have now been reported; a rapid survey based upon articles quoted by Sci-Finder Chemical Abstract Data Base (22) revealed about 300 articles published since 1999, with a steady increase; they concern new developments and applications, chiefly in water and air pollution, food and food pollution, and biological and biomedical fields 14–21).
The main advantages of SBSE and HSSE derive from the fact that these techniques are easy to apply and automate, highly flexible (the same SBSE device can be used indifferently not only for sampling in vapor or liquid phases but also for heterogeneous aqueous samples [23]), little influenced by unfavorable phase-ratio conditions, highly sensitive, robust, repeatable, reproducible and last but not least, can be applied to sample analytes with a highly different chemical structure (see the following text). On the other hand, the chief disadvantages are that it is based upon a single apolar polymer (PDMS), it only can be applied to medium-high volatility and medium-high thermo-stability analytes if a thermal desorption is to be employed, only a few solvents compatible with PDMS can be adopted for analyte liquid desorption, sampling times are long when larger volumes of PDMS are used, and the cost of instrumentation is high.
Considerable research effort has been made over recent years to overcome these limits, in particular to enable SBSE to be applied to low-volatility analytes and to increase its sampling effectiveness for polar compounds.
SBSE Sampling of Low-Volatility Analytes
At present, the recovery of low-volatility analytes is carried out by submitting SBSE devices to liquid desorption, in general followed by HPLC analysis, or by increasing their volatility by in-situ or in-tube derivatization, although in-situ derivatization has been devoted mainly to modify analyte polarity to improve their recovery in PDMS (see the following section).
SBSE–Liquid Desorption
Most SBSE applications involve the use of thermo desorption to recover the analytes accumulated in the PDMS SBSE devices in on-line combination with GC or GC–MS to analyze them; this limits its use to thermostable and volatile compounds. The most logical solution to recover low-volatility and thermolabile compounds from a PDMS SBSE device is by solvent desorption. Obviously, only those solvents in which PDMS is not soluble can be adopted; the most widely used are polar solvents, mainly acetonitrile and less used are methanol, acetone, and ethyl acetate.
Moreover, the recovery of analytes accumulated in the PDMS SBSE device by a solvent is determined strongly by the analyte's KO/W, because the less polar the analyte investigated, the less of it is transferred from the PDMS to the polar extracting solvent, but, on the other hand, the transfer of polar analytes from an aqueous matrix to PDMS is in general relatively slight. Therefore, SBSE–liquid desorption (LD) is mainly useful for nonvolatile and thermolabile compounds of medium polarity (that is, with KO/W around 102 that can be reextracted from PDMS SBSE devices with a suitable solvent with a relatively high yield.
SBSE and Recovery of Medium-to-High Polarity Analytes
Four approaches can be used alone or in combination with sample medium-to-high polarity analytes: analyte derivatization to change their polarity or volatility; modification of analyte solubility in the original matrix by varying its ionic strength, pH, or salting out; modification of PDMS polarity; and replacement of PDMS with a polar polymeric sorbent with similar operative characteristics and performance.
Derivatization and SBSE: A logical approach to overcoming the limits mentioned earlier and which extends the range of applicability of SBSE and HSSE is to modify analyte polarity by derivatizing them with reagents specific for a given group of compounds, thus, increasing their affinity to PDMS. In-situ derivatizations are carried out directly in the investigated sample before submitting it to SBSE. These reactions therefore involve analytes with organic functions whose derivatization increases their PDMS–water partition coefficient (that is, KO/W) and, as a consequence, their recovery but, at the same time, the reactions also should increase the analytes' volatility, simplifying their analysis by GC and increasing detection limits (24). The previously mentioned survey of SBSE literature (22) showed that the most widely applied in-situ reactions include derivatization of phenolic compounds with acetic acid anhydride to the corresponding acetates (25–28); derivatization of amines to the corresponding ethyl carbamates (29,30); derivatization of acids with ethyl chloroformate to the corresponding ethyl esters (30,31); acids and amines can therefore be derivatized in the same aliquot (30); and derivatization of organometallic compounds (in particular organo-tin and organo-mercury derivatives) either with sodium tetraethylborate to the corresponding ethyl derivatives (32,33) or with sodium tetra(n-propyl)borate to the corresponding propyl derivatives for speciation of native ethyl-mercury compounds (34). With some specific groups of compounds (for example, natural and synthetic estrogenic compounds), in-situ reactions can be combined with in-tube derivatizations with silylating reagents (for example, N,O-bis[trimethylsilyl]-trifluoroacetamide, BSTFA) run in vapor phase during the thermal desorption step that follows SBSE, to further increase volatility of the desorbed analytes (28).
Analysis of samples with analytes of highly different chemical structures: One of the advantages specific to SBSE is the possibility to be applied to samples consisting of analytes with highly different structures through the multishot approach introduced by Kawaguchi and colleagues in 2004 (35). This is a multi-SBSE–single desorption procedure that implies the parallel extractions of a given number of aliquots of the same sample, each submitted to different pretreatments or derivatizations, followed by the simultaneous thermodesorption of all resulting PDMS SBSE devices together from the same glass tube and the analysis by GC–single ion monitoring (SIM) MS of the different groups of target analytes all together. Multishot analysis can be applied to analyze, in a single step, samples containing compounds with widely different structures or organic functions, whose polarity is made more compatible with PDMS by specific in-situ derivatization, and whose partition coefficient is varied in favor of PDMS by modifying ionic strength, pH, or salting out the original matrix. SIM MS operated with highly selective and diagnostic ions is therefore indispensable as a GC detection method to avoid serious mistakes in analyte identification and quantitation because of peak overlapping. Recently, Sandra and colleagues have successfully applied multishot analysis to screen phenol, amine and acid, organotin, and highly apolar endocrine-disrupting chemicals (EDCs) and pharmaceuticals at the parts-per-trillion level in aqueous samples (24). Figure 2 reports the GC–SIM MS profiles of a set of target analytes representative of different groups of EDCs spiked at 100 ppt in a real-world water sample.


Figure 2
Ochiai and colleagues (36) introduced sequential SBSE to optimize sampling of ultratrace pollutants with different KO/W in water. SBSE is here performed with two stir bars sequentially applied to the same sample aliquot: the first step is done without adding modifiers with a first stir bar, the second step after salting out (30% sodium chloride) the same sample and the second stir bar. After extraction, the two stir bars are placed in the same glass desorption liner and simultaneously desorbed, and the recovered analytes are analyzed by GC–MS. With this approach, compounds with high KO/W (log KO/W > 4.0) are recovered mainly in the first extraction and those with low and medium KO/W (log KO/W <4.0) in the second. Experiments on 80 model pesticides (organochlorine, carbamate, organophosphorus, pyrethroid, and so forth) showed that sequential SBSE provides a more uniform enrichment than conventional SBSE over the entire polarity–volatility range of the model organic pollutants in water.
Sorptive Phases for Polar Compounds
Although the previously mentioned approach assures high recoveries, in particular at ultra trace levels, the most effective solution for medium-to-highly polar compounds would be to find a polar polymer with the same sorptive properties as PDMS, but at the same time with a better affinity for low KO/W compounds (that is, log KO/W <2.0). Several attempts have been made to find new materials but, at present, a polar polymer whose use is as general as that of PDMS for apolar compounds has yet to be found. On the other hand, approaches aiming at tuning the SBSE device polarity and PDMS sorptive properties have been developed successfully.
Modification of PDMS polarity: Bicchi and colleagues (37) introduced a new approach combining the concentration capabilities of two or more sampling materials operating with different mechanisms (for example, sorption and adsorption). The resulting tools for HSSE and SBSE are known as dual-phase SBSE devices; Figure 1b gives a scheme of a dual-phase SBSE device. They consists of a short PDMS tube (1–2 cm long) the ends of which are closed with two magnetic stoppers, thus creating an inner cavity that can be packed with different types of adsorbents. The concentration capability of dual-phase SBSE devices is therefore the result of the sorption of the analytes onto PDMS from liquid or vapor phase, followed by their diffusion through the PDMS layer and adsorption onto the inner phase. The effectiveness of dual-phase SBSE devices therefore depends upon the permeability of the outer PDMS layer, the adsorption capability of the inner material, and, last but not least, the intensity of the analyte–inner phase interaction; this must be reversible to afford their total release by thermal desorption. The contributions made by different PDMS and inner materials to recovery, repeatability, and intermediate precision were investigated in a long series of experiments on both standards mixtures and real-world samples, and the most effective adsorbents were found to be Carbopack (Supelco, Bellefonte, Pennsylvania) B, Tenax GC (Buchem BV, Apeldoorn, The Netherlands), a bisphenol-PDMS copolymer, and Carbopack coated with 5% of Carbowax (Supelco) (38). Figure 3 reports the GC profiles of the headspace of an arabica roasted coffee sampled by HSSE with a conventional PDMS twister and a dual-phase SBSE device with Tenax GC as inner phase. Figure 4 shows the contribution of dual-phase SBSE devices packed with carbopack B, Tenax GC, and PDMS-bisphenol copolymer as inner phases to the recovery of coffee markers compared with the corresponding empty PDMS tubing.

Figure 3
Concentration capability of PDMS also can be improved by modifying its polarity with the help of a solvent impregnating the polymer. With this approach, analytes are concentrated in an organic solvent stored inside a short piece of PDMS tubing suspended into the aqueous sample as they diffuse through the PDMS, which acts as a selective membrane leading to the inner solvent. The PDMS polarity and, hence, its solubility power is modified by the solvent diffusing through it into the aqueous sample. After a fixed time, the inside PDMS solvent containing the sampled analytes is recovered and submitted to GC or HPLC analysis. Janska and colleagues (39) introduced solvent in silicone tube extraction (SiSTEx) using acetonitrile as PDMS modifier in combination with GC–MS to analyze 26 pesticides at parts-per-billion level. Van Hoeck and colleagues (40) applied silicon membrane sorptive extraction (SMSE), using ethyl acetate as PDMS modifier in combination with GC–SIM MS to quantify atrazine (ATR) and its three metabolites in water samples in the 1–10 ppt range. Figure 1c shows a diagram of a SMSE sampling device.

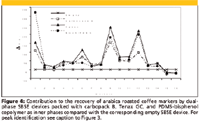
Figure 4
SMSE also has been applied to headspace sampling by Sgorbini and colleagues (41). In this case, the solvent diffusing through the PDMS modifies its polarity but the analytes are accumulated into the polymer, from where they are recovered by thermal desorption followed by GC or GC–MS analysis. Headspace SMSE was applied successfully to headspace sampling of sage and thyme with ethyl acetate and cyclohexane as PDMS modifiers, coffee with PDMS–ethyl acetate, and fatty acids with PDMS–ethyl acetate at pH 8.
Sorptive extraction with polar sorbent: Several materials with different polarity have been proposed to replace PDMS as coating for stir bars, but most of them were developed mainly (or were only effective) to sample specific groups of analytes selectively. Among others, Lambert and colleagues (42) developed a robust and effective biocompatible stir bar coated with alkyl-diol-silica (ADS) restricted access material (RAM) as coating for the direct extraction of caffeine and metabolites in biological fluids. Sampled analytes were recovered by solvent desorption (3:1 [v/v] water–acetonitrile) and analyzed by HPLC with UV absorbance detection.
Zhu and colleagues developed stir bars coated with a molecularly imprinted polymer (MIP) consisting of a film formed from a formic acid solution of nylon-6 polymer, either nonimprinted or imprinted with monocrotophos to extract successfully and selectively the four structural analogs of monocrotophos from dichloromethane solution for the analysis of environmental and biolological samples (MIP film thickness: 180 μm) (43) and with L-glutamine to extract this amino acid (MIP film thickness: 160 μm) (44). Huang and colleagues (45–47) developed a number of coatings based upon monolithic material. The first one was obtained by in-situ copolymerization of octyl methacrylate and ethylene dimethacrylate using a 1-propanol, 1,4-butanediol based porogen solvent and water with azo-bis-isobutyronitrile as initiator. The resulting stir bars were applied successfully to the polycyclic aromatic hydrocarbon (PAH) determination in seawater samples (45). Other monolithic materials were synthesized to improve the performance with polar compounds: a poly-methacrylic acid stearyl ester-ethylene dimethacrylate derivative that was used to analyze steroid sex hormones as model molecules and in urine samples (46) and a modification of it (polyvinylpyridine-ethylene dimethacrylate) to analyze strongly polar phenols in water and in lake and sea water (47). In all cases, analytes were recovered from the stir bar by solvent desorption and analyzed by HPLC.
More recently, Nogueira and colleagues (48–50) synthesized and tested several polyurethane foams to be applied as stir bar coating material and showed their effectiveness in the enrichment of highly polar metabolites from water matrices, thus, overcoming the limitations of the conventional PDMS phase. In a first group of experiments, polyurethane foams were shown to give high recoveries for atrazine, 2,3,4,5-tetrachlorophenol, and fluorene, used as model compounds, in water samples at a trace level, even in comparison to conventional PDMS SBSE devices; they also showed remarkable stability and excellent mechanical resistance to organic solvents (48). Recovery of sampled analytes from the stir bar was by solvent desorption and analysis not only by HPLC but also by large volume injection–GC–MS. A different polyurethane foam was applied successfully to sample seven triazinic herbicides (simazine, atrazine, prometon, ametryn, propazine, prometryn, and terbutryn) in water matrices and was used to analyze real-world samples of ground and surface waters; results were good including when compared with those obtained using conventional PDMS SBSE devices. Liquid desorption by methanol was used to recover the sampled analytes (49). Polyurethane foam stir bars also were applied successfully to the analysis of the residues of six acidic pharmaceuticals (o-acetylsalicylic acid, ibuprofen, diclofenac sodium, naproxen, mefenamic acid, and gemfibrozil), selected as model compounds of nonsteroidal acidic anti-inflammatory drugs and lipid regulators in environmental water matrices, including river, sea, and wastewater samples (50).
Conclusions
Over the last few years, SBSE and HSSE have enjoyed an ever-increasing success for ultratrace analysis in water and air pollution, food and food pollution, and biological and biomedical fields because of the methods' high reliability. Recent developments have widely extended their use to polar and thermally unstable analytes, although a considerable research effort has still to be made to find (or develop) highly effective polymeric material for polar compounds with sorptive performance similar to those of PDMS.
Acknowledgment
The authors are indebted to the project entitled: "Sviluppo di metodologie innovative per l'analisi di prodotti agroalimentari" (FIRB Cod.: RBIP06SXMR_002) of the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) (Italy).
Ronald E. Majors "Sample Prep Perspectives" Editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, DE, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com
References
(1) F. David, B. Tienpont, and P. Sandra, LCGC 21(2), 108–118 (2003).
(2) E. Baltussen, P. Sandra, F. David, and C. Cramers, J. Microcol. Sep. 11, 737–747 (1999).
(3) P. Popp, C. Bauer, and L. Wennrich, Anal. Chim. Acta 436(1), 1–9 (2001).
(4) P. Sandra, B. Tienpont, J. Vercammen, A. Tredoux, T. Sandra, and F. David, J. Chromatogr., A 928(1), 117–126 (2001).
(5) C. Bicchi, C. Cordero, C. Iori, P. Rubiolo, and P. Sandra, J. High Resolut. Chromatogr. 23, 539–546 (2000).
(6) B. Tienpont, F. David, C. Bicchi, and P. Sandra, J. Microcol. Sep. 12, 577–584 (2000).
(7) E. Baltussen, F. David, P. Sandra, H.G. Janssen, C. and Cramers, J. High Resolut. Chromatogr. 21, 332–340 (1998).
(8) E. Baltussen, P. Sandra, F. David, H.G. Janssen, C. and Cramers, Anal. Chem. 71, 5213–5216 (1999).
(9) E. Baltussen, PhD Thesis, Technische Universireit Eindhoven (The Netherlands), Sept. 11, 2000, p. 10.
(10) E. Baltussen, C. Cramers, and P.J.F. Sandra, Anal. Bioanal. Chem. 373(1–2), 3–22 (2002).
(11) F. David and P. Sandra, J. Chromatogr., A 1152(1–2), 54–69 (2007).
(12) T. Hyotylainen, M.L. Riekkola, Anal. Chim. Acta 614(1), 27–37 (2008).
(13) C. Bicchi, C. Cordero, E. Liberto, B. Sgorbini, and P. Rubiolo, J. Chromatogr., A 1184, 220–233 (2008).
(14) M. Kawaguchi, Hoshi Yakka Daigaku Kiyo, 48, 43–50 (2006).
(15) M. Kawaguchi, R. Ito, K. Saito, and H. Nakazawa, J. Pharm. Biomed. Anal. 40(3), 500–508 (2006).
(16) A. Nongonierma, P. Cayot, J.-L. Le Quere, M. Springett, and A. Voilley, Food Rev. Int. 22(1), 51–94 (2006).
(17) J.B. Quintana and I. Rodriguez, Anal. Bioanal. Chem. 384(7–8), 1447–1461 (2006).
(18) Y. Pico, M. Fernandez, M. J. Ruiz, and G. Font, J. Biochem. Biophys. Meth. 70(2), 117–131 (2007).
(19) A. Mosandl, M. Kreck, J. Jung, S. Sewenig, ACS Symposium Series 952, 52–74 (2007).
(20) K. Ridgway, S.P.D. Lalljie, and R.M. Smith, J. Chromatogr., A 1153(1–2), 36–53 (2007).
(21) R. Castro, R. Natera, E. Duran, and C. Garcia-Barroso, Eur. Food Res. Technol. 228(1), 1–18 (2008).
(22) Sci-Finder Chemical Abstract Data Base, America Chemical Society, Washington , DC, http://www.cas.org.
(23) C. Bicchi, C. Cordero, P. Rubiolo, P. Sandra, Eur. Food Res. Technol. 216, 449–456 (2003).
(24) E. Van Hoeck, F. Canale, C. Cordero, S. Compernolle, C. Bicchi, and P. Sandra, Anal. Bioanal. Chem., (2008), DOI 10.1007/s00216-008-2339-7.
(25) M. Kawaguchi, Y. Ishii, N. Sakui, N. Okanouchi, R. Ito, K. Inoue, K. Saito, and H. Nakazawa, J. Chromatogr., A 1049, 1–8 (2004).
(26) N. Itoh, H. Tao, and T. Ibusuki, Anal. Chim. Acta 535, 243–250 (2005).
(27) M. Kawaguchi, Y. Ishii, N. Sakui, N. Okanouchi, R. Ito, K. Saito, and H. Nakazawa, Anal. Chim. Acta 533(1), 57–65 (2005).
(28) M. Kawaguchi, R. Ito, N. Sakui, N. Okanouchi, K. Saito, and H. Nakazawa, J. Chromatogr., A 1105, 140–147 (2006).
(29) H. Kataoka, J. Chromatogr., A 733, 19–34 (1996).
(30) B.Tienpont, F. David, K. Desmet, and P. Sandra, Anal. Bioanal. Chem. 373, 46–55 (2002).
(31) P. Husek, J. Chormatogr., B 717, 57–91 (1998).
(32) C. Devos, M. Vliegen, B. Willaert, F. David, L. Moens, and P. Sandra, J. Chromatogr., A 1079, 408–414 (2005).
(33) A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria, L.A. Fernandez, C. Marcic, A. de Diego, J. Chromatogr., A 1185(1), 130–138 (2008).
(34) R. Ito, M. Kawaguchi, N. Sakui, H. Honda, N. Okanouchi, K. Saito, and H. Nakazawa, J. Chromatogr., A 1209 (1–2), 267–270 (2008).
(35) M. Kawaguchi, Y. Ishii, N. Sakui, N. Okanouchi, R. Ito, K. Inoue, K. Saito, and H. Nakazawa, J. Chromatogr., A 1049, 1–8 (2004).
(36) N. Ochiai, K. Sasamoto, H. Kanda, and E. Pfannkoch, J. Chromatogr., A 1200(1), 72–79 (2008).
(37) C. Bicchi, C. Cordero, E. Liberto, P. Rubiolo, B. Sgorbini, F. David, and P. Sandra, J. Chromatogr., A 1094(1–2), 9–16 (2005).
(38) C. Bicchi, C. Cordero, E. Liberto, B. Sgorbini, F. David, P. Sandra, and P. Rubiolo, J. Chromatogr., A 1164(1–2), 33–39 (2007).
(39) M. Jánská, S. J. Lehotay, K. Maštovská, J. Hajšlová, T. Alon, and A. Amirav, J. Sep.Sci. 29, 66–80 (2006).
(40) E. Van Hoeck, F. David, and P. Sandra, Chromatographia in press.
(41) B. Sgorbini, D. Budziak, C. Cordero, E. Liberto, P. Rubiolo, P. Sandra, and C. Bicchi, 32nd Int. Symp. on Capillary Chromatography, May 26-30, 2008, Riva del Garda, (Italy) poster C21, p 167.
(42) J.-P. Lambert, W.M. Mullett, E. Kwong, and D. Lubda, J. Chromatogr., A 1075(1–2), 43–49 (2005).
(43) X. Zhu, J. Cai, J. Yang, Q. Su, and Y. Gao, J. Chromatogr., A 1131(1–2), 37–44 (2006).
(44) X. Zhu and Q. Zhu, J. Appl. Polymer Sci. 109(4), 2665–2670 (2008).
(45) X. Huang and D. Yuan, J. Chromatogr., A 1154(1–2), 152–157 (2007).
(46) X. Huang, D. Yuan, J.B.L. Huang, Talanta 75, 172–177 (2008).
(47) X. Huang and D. Yuan, J. Chromatogr., A 1194(1), 134–138 (2008).
(48) N.R. Neng, M.L. Pinto, J. Pires, P. M. Marcos, J.M.F. Nogueira, J. Chromatogr., A 1171(1–2), 8–14 (2007).
(49) F.C.M. Portugal, M.L. Pinto, and J.M.F. Nogueira, Talanta 77(2), 765–773 (2008).
(50) A.R.M. Silva, C.M. Portugal, and J.M.F. Nogueira, J. Chromatogr., A 1209(1–2), 10–16 (2008).






 and Headspace Sorptive Extraction (HSSE): An Overview")

Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.


